

In this issue of Blood, identify a mechanism that couples the treatment of acute ischemic stroke (AIS) with tissue plasminogen activator (tPA) to the development of intracerebral hemorrhage (ICH). Their findings suggest that treatment with tPA results in neutrophil recruitment, activation, and neutrophil extracellular trap (NET) formation that provokes the breakdown of the blood-brain barrier (BBB) and hemorrhage through a cyclic GMP-AMP (c-GAMP) synthase-stimulator of interferon genes (cGAS-STING) signaling pathway and a type I interferon (IFN) response in the ischemic brain.1
Stroke is the fifth leading cause of death in the United States, with AIS accounting for 87% of all cases.2 Indeed, it is estimated that someone in the United States dies as a result of stroke every 4 minutes. Stroke accounts for an approximate cost to the US health care system of $45 billion.2 In 1996, the National Institute of Neurological Disorders and Stroke tPA trial demonstrated the efficacy of intravenous recombinant tPA (r-tPA), which improved patient outcomes if administered within 3 hours of the ischemic event. Today, r-tPA remains the only approved pharmacologic therapy for AIS. Despite the benefits of tPA therapy, there is significant risk because of an increased frequency of symptomatic ICH, which occurs in 6% to 7% of patients treated with r-tPA. Development of ICH in AIS patients treated with r-tPA is catastrophic, with a mortality rate of up to 83%.3 Thus, improving our understanding of the mechanisms that lead to ICH after treatment with r-tPA and identifying novel therapeutic targets is a significant priority for improving the clinical management of stroke.
The role of tPA in fibrinolysis is well established. The tPA serine protease is synthesized primarily by endothelial cells and hepatocytes and circulates at low levels in an inactive form.4 The activity of tPA is significantly upregulated after binding to fibrin matrices, which mediates local conversion of the zymogen serine protease plasminogen to the active fibrinolytic enzyme plasmin. Conceptually, this mechanism is well-suited for the use of tPA as a thrombolytic agent. However, when delivered as an intravenous bolus in cases of stroke (and myocardial infarction), the activity of tPA is not limited to plasminogen activation on fibrin matrices. At therapeutic concentrations, tPA can drive plasmin(ogen) activation when bound to circulating fibrinogen, thus mediating fibrinogenolysis, which results in fibrinogen consumption and reduced hemostatic potential. Moreover, tPA has molecular and cellular activities independent of plasminogen activation and fibrinolysis.
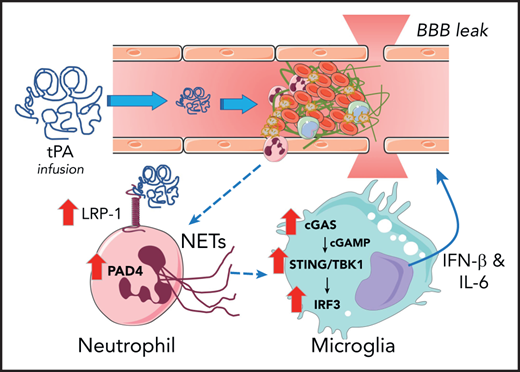
Model by which ICH occurs in stroke after tPA infusion therapy. Introduction of therapeutic concentrations of tPA results in upregulation of LRP-1 and PAD4 by neutrophils, which results in induction of NETosis. Neutrophil-derived DNA results in activation of the cGAS-STING/TNKB1/IRF3 pathway, which promotes the release of type I interferon and IL-6 that cause BBB leak.
Model by which ICH occurs in stroke after tPA infusion therapy. Introduction of therapeutic concentrations of tPA results in upregulation of LRP-1 and PAD4 by neutrophils, which results in induction of NETosis. Neutrophil-derived DNA results in activation of the cGAS-STING/TNKB1/IRF3 pathway, which promotes the release of type I interferon and IL-6 that cause BBB leak.
Studies have documented that administration of tPA can have multiple unintended pathologic consequences in neuronal tissue, including neuronal cell death, enhanced BBB permeability, and cerebrovascular inflammation. BBB permeability mediated by tPA was shown to be a plasminogen-independent mechanism regulated through the low-density lipoprotein receptor-related protein-1 (LRP-1).5 In animal models of stroke, tPA can enhance neutrophil recruitment, accumulation, and activation after the ischemic event, and this neutrophil recruitment can exacerbate the hemorrhagic transformation. Over the last decade, neutrophils and neutrophil function have been linked to a number of pathologies primarily because of their ability to release NETs, which are long strands of extracellular DNA, histones, and other proteins.6 NETs can provoke thrombosis, inflammation, tissue damage, and tissue disrepair. Importantly, histologic evidence of neutrophils and NETs has been found in clots retrieved from patients with AIS by mechanical thrombectomy.7
The Wang et al study highlights that in a middle cerebral artery occlusion (MCAO) model of AIS in mice, tPA infusion substantially enhanced the accumulation of neutrophils, increased local levels of the neutrophil enzyme myeloperoxidase, and increased the NET marker citrullinated histone H3 (H3Cit) in the infarcted ischemic cortex. Ex vivo studies of neutrophils harvested from MCAO-challenged and sham-operated controls indicated that ischemia sensitized neutrophils to tPA stimulation, which resulted in elevation of H3Cit and NET formation. The mechanism of tPA-mediated NET formation was linked to expression of LRP-1 by neutrophils and subsequent upregulation of protein arginine deiminase-4 (PAD4), an enzyme required for NET formation. Inhibition of LRP-1 by the antagonist receptor-associated protein (RAP) blocked both PAD4 upregulation and NET formation. The functional contribution of the tPA-neutrophil-LRP-1-PAD4 axis to tPA-induced ICH after ischemic stroke was shown. DNase I treatment (to degrade NETs) or genetically imposed PAD4 deficiency (to prevent NET formation) mitigated reactive changes in the brain after ischemic challenge. Notably, both interventions significantly suppressed tPA-driven BBB permeability, mitigated loss of gap junction proteins (ie, zonula occludens-1 [ZO-1], occludin, claudin-5, and vascular endothelial-cadherin [VE-cadherin]), reduced ICH, and improved neurologic deficits (as shown by behavioral testing).
The authors identify a pathway whereby the release of DNA by neutrophils leads to the activation of the cytosolic DNA sensor cGAS in microglial cells and macrophages within the ischemic brain. The cGAS sensor promotes the generation of cGAMP and downstream activation of the STING/TANK-binding kinase-interferon regulatory pathway-3 (TBK1-IRF3) pathway that induces IFN-β and interleukin-6, leading to BBB leak (see figure). This pathway was rigorously analyzed in vivo through both loss-of-function (eg, cGAS deficiency) and gain-of-function (eg, administration of cGAMP) approaches. The identification of microglial cells as being a key player is of interest because microglial cells, the cGAS-STING pathway, and neutrophils have each been identified as mediators of tissue damage in other neuroinflammatory and cerebrovascular diseases such as multiple sclerosis and Alzheimer’s disease.8-10 Thus, it would be of interest to examine whether this same axis contributes to neural degeneration in those diseases and whether endogenous tPA contributes to exacerbating disease.
The findings provide an important reminder that despite the utility of using tPA as a thrombolytic, potential off-target effects must be carefully considered when it is used to treat AIS. These studies provide a framework and highlight a pathway forward for designing and testing adjuvant therapies to improve the safety profile of tPA for thrombolysis in AIS. The authors have identified candidates (eg, DNase I and RAP) and putative druggable targets (eg, PAD4, cGAS, STING) for alleviating ICH triggered by tPA infusion therapy for stroke. However, just as tPA treatment provokes deleterious pathways, it will be important to experimentally determine whether such adjuvant therapies result in unintended consequences, including altering coagulation or fibrinolytic pathways in ways that negatively impact thrombolysis.
Conflict-of-interest disclosure: The author declares no competing financial interests.


