

In this issue of Blood, reveal that the RNA-binding protein YBX1 is essential for acute myeloid leukemia (AML) development and propagation but is not critical for normal hematopoietic stem cell (HSC) functions and multilineage hematopoiesis.1 Mechanistically, the authors propose that YBX1 promotes AML by interacting with IGF2BPs, readers of messenger RNAs (mRNAs) that are chemically modified by methylation at the N6 position of adenosines (m6A), to stabilize these transcripts. Among these stabilized transcripts are those encoding MYC and BCL2, key regulators of AML cell proliferation and survival.
Given that long-standing therapeutic strategies in AML often fail to fully eliminate leukemic stem cells that fuel disease and are ultimately responsible for fatal relapses, the AML community has turned to new fields in search of novel efficient therapeutic targets. Although the role of gene transcription in leukemogenesis has been studied extensively, the functional significance of posttranscriptional regulation of gene expression, including RNA modifications, is only beginning to emerge. Recent evidence indicates that m6A, the most abundant internal mRNA modification, is an important regulator of normal and malignant hematopoiesis.2-7 The m6A modification is installed in the proximity of stop codons and 3′ untranslated regions by the m6A methyltransferase complex (“m6A writer”) and can be removed by m6A demethylases (collectively called “m6A erasers”).8 The functions of the m6A modification are predominantly executed by YTH domain-containing m6A readers (including nuclear YTHDC1, and cytoplasmic readers YTHDF1-3 and YTHDC2). Although nuclear YTHDC1 regulates mRNA splicing and nuclear export, cytoplasmic m6A readers collectively promote decay and translation of m6A-modified mRNAs. Furthermore, a recently discovered class of cytosolic readers, insulin-like growth factor 2 mRNA-binding proteins 1-3 (IGF2BP1-3), functions to stabilize m6A-modified mRNAs.9 Significantly, m6A writers and erasers are overexpressed in AML, and their inactivation cripples AML cells through multiple m6A-dependent mechanisms.4-7 Furthermore, the mRNA m6A reader YTHDF2 is highly expressed across human AML, and its inactivation selectively compromises AML development and propagation by extending the half-life of m6A-modified transcripts, including TNFR2, thus sensitizing them to apoptosis.3 Despite promising emerging evidence of the therapeutic potential of m6A mRNA modification in AML, the functional significance of other m6A readers, including IGF2BPs, and their regulators, has remained elusive.
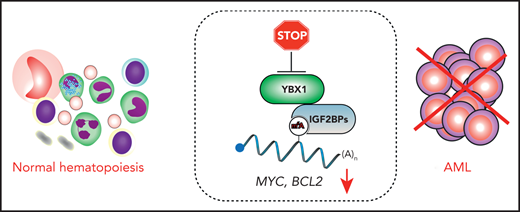
YBX1 interacts with the m6A reader IGF2BP proteins to stabilize m6A-modified transcripts, including MYC and BCL2. Upon YBX1 inactivation, MYC and BCL2 undergo accelerated decay, thus compromising AML cells. However, the loss of YBX1 has no major impact on normal multilineage hematopoiesis.
YBX1 interacts with the m6A reader IGF2BP proteins to stabilize m6A-modified transcripts, including MYC and BCL2. Upon YBX1 inactivation, MYC and BCL2 undergo accelerated decay, thus compromising AML cells. However, the loss of YBX1 has no major impact on normal multilineage hematopoiesis.
Feng et al sought to determine the role of YBX1, a known interactor of IGF2BPs, in AML pathogenesis. They found that YBX1 was overexpressed in human AML cells with normal karyotype and various different cytogenetic subtypes. Furthermore, YBX1 knockdown in human AML cells impaired their survival and disease propagation capacity. To determine the requirement for YBX1 in AML development, the authors genetically deleted Ybx1 in mouse hematopoietic stem/progenitor cells (HSPCs) prior to their transformation with MLL-AF9 oncogene and revealed that HSPCs lacking Ybx1 failed to efficiently establish AML. To investigate the significance of YBX1 in AML propagation, they acutely knocked down the expression of Ybx1 in established AML cells driven by MLL-AF9 and discovered that they were unable to successfully propagate AML in vivo. Given the finding that YBX1 was required for AML initiation and maintenance, Feng et al investigated the impact of YBX1 loss on HSC functions and normal multilineage hematopoiesis. Surprisingly, Ybx1 was not essential for the maintenance of HSCs and progenitor cells at different levels of the differentiation hierarchy and multilineage hematopoiesis. Together, these results position YBX1 as a promising therapeutic target in AML, whose inhibition is likely to compromise AML cells while leaving normal hematopoiesis mostly intact (see figure). However, to confirm a therapeutic window for targeting YBX1, it would be of major importance to examine the impact of YBX1 inactivation on other healthy tissues. Finally, although this study largely focused on AML with MLL translocation, further studies are needed to determine whether targeting YBX1 could be of clinical utility in other more frequent AML subtypes.
Understanding the molecular mechanisms of YBX1’s functions in AML is a prerequisite for designing efficient strategies for its potential future therapeutic targeting. Feng et al found that YBX1 binds m6A-modified transcripts indirectly through physical interactions with m6A readers in the IGF2BP family, which bind m6A mRNA through the UGGAC consensus sequence containing the core m6A motif. Interestingly, RNA immunoprecipitation coupled to sequencing revealed that YBX1 and IGF2BPs cooccupy largely overlapping m6A-modified mRNAs, including transcripts known to promote AML cell survival and proliferation, such as MYC and BCL2. Mechanistically, the m6A readers IGF2BP1-3 recognize m6A-modified transcripts and enhance mRNA stability.9 Importantly, Feng et al revealed that IGF2BPs require YBX1 to promote m6A mRNA stability, as YBX1 loss significantly accelerated m6A mRNA decay, including those encoding MYC and BCL2. Finally, in a series of elegant rescue experiments, the authors demonstrated that ectopic expression of MYC and BCL2 partially rescued AML survival and proliferation defects resulting from YBX1 inactivation. Therefore, YBX1 promotes stability of m6A-modified transcripts required for AML cell propagation by interacting with m6A readers IGF2BP1-3. To exert its functions in AML, YBX1 requires binding to IGF2BPs; however, it remains to be determined whether the YBX1-IGF2BP interaction can be pharmacologically disrupted for therapeutic purposes. Another therapeutic opportunity could be to inhibit the binding of IGF2BPs proteins to m6A. Before this strategy is implemented, it would be of immense interest to elucidate the role of IGF2BP1-3 in leukemogenesis and normal hematopoiesis and investigate possible redundancies between these m6A readers.
Although Feng et al revealed that YBX1 promotes m6A mRNA stability via IGF2BP1-3 in AML, we know that it also binds YTH domain-containing m6A readers, including YTHDF2,1 which is required for AML propagation,3 indicating that YBX1 may have a broader impact on the metabolism of m6A-modified transcripts. This fundamental issue certainly merits further investigation. It is also important to note that YBX1 has pleiotropic and diverse functions beyond m6A, acting as a reader of 5-methylcytosine RNA modification and regulating splicing. Indeed, a recent report revealed that YBX1 functions downstream of JAK2-V617F mutation in myeloproliferative neoplasms to promote efficient mRNA splicing, and YBX1 inactivation results in intron retention, thereby sensitizing cells to apoptosis.10 In conclusion, YBX1 has emerged as a promising target in blood malignancies, and understanding the mechanisms through which it functions will inform strategies for most efficient therapeutic development.
Conflict-of-interest disclosure: The author declares no competing financial interests.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal