

In this issue of Blood, report that constitutive activation of AKT is common in high-risk chronic lymphocytic lymphoma (CLL) and Richter transformation (RT), and demonstrate that genetic constitutive activation of AKT in B cells in the TCL1 mouse model of CLL drives transformation to an aggressive lymphoma.1 RT is the development of an aggressive lymphoma in the background of CLL, most typically diffuse large B-cell lymphoma, estimated to occur in 2% to 10% of patients. RT remains the greatest unmet need in CLL therapy in the era of targeted therapies, with poor response to treatment and very short overall survival, estimated at only 3.3 months in a recent multicenter retrospective study.2 Although some risk factors for developing RT are known, and include CLL with aberrant TP53, unmutated IGHV, NOTCH1 mutation, and stereotyped B-cell receptors,3 nonetheless most patients whose CLL has these features will not develop RT. Given the markedly worse outcome once RT develops,2 a major goal for the field must be to better predict which patients will develop this catastrophic event, and prevent it.
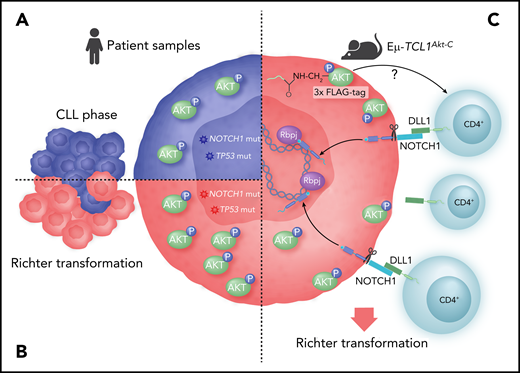
Modeling the role of AKT in RT. (A) CLLs with high-risk features show constitutive activation of AKT (pAKT). (B) More than one-half of RTs show constitutive activation of AKT. (C) Genetic constitutive activation of AKT in the TCL1 mouse model leads to expansion of microenvironment CD4 cells expressing the NOTCH ligand DLL1, leading to activation of NOTCH1, which drives transformation.
Modeling the role of AKT in RT. (A) CLLs with high-risk features show constitutive activation of AKT (pAKT). (B) More than one-half of RTs show constitutive activation of AKT. (C) Genetic constitutive activation of AKT in the TCL1 mouse model leads to expansion of microenvironment CD4 cells expressing the NOTCH ligand DLL1, leading to activation of NOTCH1, which drives transformation.
In that context, the work of Kohlhaas et al is exciting. They find that constitutive phosphorylation of AKT is present in more than one-half of RT samples studied, and high among CLLs at high risk for RT, namely those with NOTCH1 mutation, TP53 mutation, or 17p deletion (see figure). The levels of pAKT appear uniformly increased in these CLL subsets, with the caveat of small numbers tested, whereas patients with lower genomic risk show greater variability, suggesting a possible biomarker for RT risk. These findings justify larger patient studies for validation.
To determine whether this AKT activity is sufficient to transform CLL to RT, the authors express a constitutively active Akt allele (Akt-C) in B cells in the TCL1 mouse model of CLL. Although the TCL1 mice with and without Akt-C have similar progression of a CLL-like disease early on, by about 7 months of age, the mice with Akt-C develop an aggressive lymphoma with massive splenomegaly and significantly reduced overall survival, demonstrating that constitutively active AKT does drive a RT-like transformation in this mouse model. These results are in interesting contrast to what is observed with B-cell-specific knockout of Trp53 or Atm in the TCL1 mouse, which results in a more aggressive CLL-like phenotype, but only occasional transformation.4
To understand the transformation, the authors characterize the observed RT in comparison with the CLL-like disease in the TCL1 mice. Interestingly, at a genomic level, the mutational pattern appears intermediate between CLL and diffuse large B-cell lymphoma, with a similar mutation burden to the CLL and without recurrently mutated genes. Although genomic data on patient RT are limited, they do suggest about 20 newly acquired genetic lesions per case,5 more than in this model. However, using phospho-proteomics and single-cell RNA sequencing, the authors observe increased NOTCH activation and signaling uniquely in the TCL1 mice with Akt-C and trace this activation to an expansion of CD4 T cells in the microenvironment (see figure). These CD4 T cells show upregulation of the NOTCH1 ligand DLL1 on their surface, which then activates NOTCH1 on the tumor cells, presumably driving transformation. How pAKT drives CD4 T-cell expansion in the microenvironment is an interesting unanswered question.
To further confirm that Akt-C induces transformation through NOTCH1 activation, the authors cross a constitutively active Notch1 allele into the TCL1 mouse background. Interestingly, these mice do not develop disease early on; in fact, they have no mortality until the same sudden onset of massive splenomegaly with aggressive lymphoma morphology that was seen in the TCL1Akt-C mice. The timing is significantly delayed compared with the TCL1Akt-C mice, suggesting that constitutive activation of AKT is significantly accelerating the phenotype compared with NOTCH1 activation alone. Thus, constitutive AKT may amplify the NOTCH1 signal or add additional signalsw that accelerate transformation. These findings have potential implications for patients with NOTCH1 mutated CLL, providing additional evidence of their increased risk of transformation and suggesting that constitutive AKT activation in the CLL phase may confer additional risk.
What then are the clinical implications of these findings? In many ways, they raise more questions than answers. For example, how well does the TCL1 mouse model mimic human CLL, and in that context, how well does this model mimic RT in patients? The model uses a genetically constitutively active Akt allele, which is rarely seen in CLL or RT, and may result in quantitatively or even qualitatively different activation than more typical pathway stimulation. AKT activation should be evaluated in other mouse models of RT, including the Eμ-TCL1xMyc model6 and PDX models,7 as well as more extensively in patient RT. These findings also justify intensive research into pAKT as a potential biomarker of RT risk, in both high and low genomic risk CLL patients, before and during therapy. For example, a small subset of patients usually with TP53-aberrant CLL still develop transformation soon after starting BTK inhibitor therapy,8 and AKT activation before or after therapy initiation should be tested as a possible biomarker to identify those patients early.
Constitutive AKT activation also provides an obvious therapeutic target in high-risk CLL or RT. Isoform-specific inhibitors of phosphoinositol-3-kinase (PI3K) that downregulate pAKT are already approved in CLL. The combination of the PI3Kδ,γ inhibitor duvelisib with the BCL-2 inhibitor venetoclax has shown promise in RT PDX models,9 and a multicenter trial in CLL and RT is ongoing (NCT03534323). Both duvelisib (NCT03892044) and the pan-PI3K inhibitor copanlisib (NCT03884998) are being separately tested in combination with nivolumab for RT. Promising preliminary data in RT have been reported with the PI3Kδ inhibitor umbralisib in combination with pembrolizumab and the anti-CD20 antibody ublituximab.10 Exportin-1 inhibitors also have activity against AKT among other pathways and have shown promise in animal models.6 The work reported here validates these ongoing studies and provides a rich set of new questions to investigate in the ongoing effort to reduce the threat of RT to our CLL patients.
Conflict-of-interest disclosure: J.R.B. has served as a consultant for Abbvie, Acerta, Astra-Zeneca, Beigene, Catapult, Juno/Celgene, Kite, MEI Pharma, Morphosys AG, Nextcea, Novartis, Octapharma, Pfizer, Rigel, Sunesis, TG Therapeutics, and Verastem; received honoraria from Janssen; received research funding from Gilead, Loxo, Sun, and Verastem; and served on data safety monitoring committees for Invectys.

