Key Points
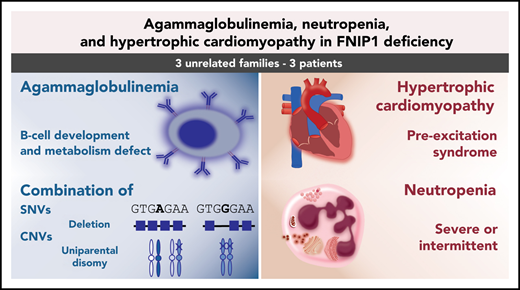
FNIP1 deficiency causes agammaglobulinemia, variable neutropenia, and hypertrophic cardiomyopathy.
FNIP1 deficiency alters B-cell development and metabolism.

Abstract
Agammaglobulinemia is the most profound primary antibody deficiency that can occur due to an early termination of B-cell development. We here investigated 3 novel patients, including the first known adult, from unrelated families with agammaglobulinemia, recurrent infections, and hypertrophic cardiomyopathy (HCM). Two of them also presented with intermittent or severe chronic neutropenia. We identified homozygous or compound-heterozygous variants in the gene for folliculin interacting protein 1 (FNIP1), leading to loss of the FNIP1 protein. B-cell metabolism, including mitochondrial numbers and activity and phosphatidylinositol 3-kinase/AKT pathway, was impaired. These defects recapitulated the Fnip1−/− animal model. Moreover, we identified either uniparental disomy or copy-number variants (CNVs) in 2 patients, expanding the variant spectrum of this novel inborn error of immunity. The results indicate that FNIP1 deficiency can be caused by complex genetic mechanisms and support the clinical utility of exome sequencing and CNV analysis in patients with broad phenotypes, including agammaglobulinemia and HCM. FNIP1 deficiency is a novel inborn error of immunity characterized by early and severe B-cell development defect, agammaglobulinemia, variable neutropenia, and HCM. Our findings elucidate a functional and relevant role of FNIP1 in B-cell development and metabolism and potentially neutrophil activity.
Introduction
Agammaglobulinemia is the most profound primary antibody deficiency that results from early termination of B-cell development, which leads to the absence of mature circulating B cells and very low or absent serum immunoglobulin levels. To date, defects in BTK, IGHM, IGLL1, CD79A, CD79B, BLNK, and PIK3R1 have been reported to cause agammaglobulinemia.1
Disruption of folliculin interacting protein 1 (FNIP1) alters the essential metabolic regulators AMPK and mTOR (Figure 1A), resulting in profound B-cell deficiency and decreased natural killer (NK) T cells, hypertrophic cardiomyopathy (HCM), and pre-excitation syndrome.2-8 Contemporarily, another group9 clinically described 2 families with inborn FNIP1 deficiency with hypogammaglobulinemia, intermittent neutropenia, and HCM. Here, we present functional validation of FNIP1 deficiency in 3 novel families, including the first adult case. FNIP1 deficiency results in HCM, absent circulating B cells, agammaglobulinemia, and either severe or intermittent neutropenia.
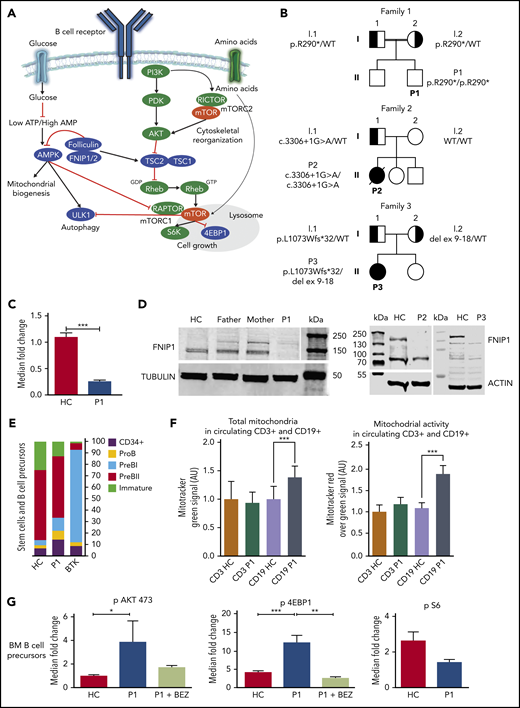
Functional studies on patients with FNIP1 deficiency. (A) Schematic illustration displaying the interaction of FNIP1 in B cells. Positive and negative regulators of mTORC1 signaling are depicted in green and in blue, respectively. Folliculin and Fnip1/2 have been described as both positive and negative regulators of mTORC1. (B) Pedigrees showing 3 families with affected individuals harboring FNIP1 variants. Solid symbols indicate affected persons who were homozygous or compound heterozygous for the mutant alleles; half solid symbols, heterozygous persons; circles, female family members; square, male family members; double lines, consanguinity. (C) Expression of FNIP1 messenger RNA in P1 (quantitative reverse-transcription polymerase chain reaction analysis). Data are expressed as mean ± standard deviation (2 independent experiments, each performed in triplicate). Statistical analysis was performed using 1-way analysis of variance. (D) FNIP1 protein expression in T cells. (E) Bone marrow B-cell immunophenotyping in P1 compared with a healthy control and 1 Bruton tyrosine kinase (BTK) patient (representative experiment). (F) Quantification of total mitochondrial abundance and mitochondrial activity in circulating CD19+ cells isolated from an healthy control and P1 (representative experiment). AU, arbitrary units. (G) Evaluation of pAKT, pS6, and p4EBP1 levels in B-cell bone marrow progenitors from P1 and a healthy control (2 experiments). In all graphs, **P < .01 and ***P < .001. Data are presented as means ± standard deviation. AMP, adenosine 5′-monophosphate; ATP, adenosine triphosphate; HC, healthy control; WT, wild type.
Functional studies on patients with FNIP1 deficiency. (A) Schematic illustration displaying the interaction of FNIP1 in B cells. Positive and negative regulators of mTORC1 signaling are depicted in green and in blue, respectively. Folliculin and Fnip1/2 have been described as both positive and negative regulators of mTORC1. (B) Pedigrees showing 3 families with affected individuals harboring FNIP1 variants. Solid symbols indicate affected persons who were homozygous or compound heterozygous for the mutant alleles; half solid symbols, heterozygous persons; circles, female family members; square, male family members; double lines, consanguinity. (C) Expression of FNIP1 messenger RNA in P1 (quantitative reverse-transcription polymerase chain reaction analysis). Data are expressed as mean ± standard deviation (2 independent experiments, each performed in triplicate). Statistical analysis was performed using 1-way analysis of variance. (D) FNIP1 protein expression in T cells. (E) Bone marrow B-cell immunophenotyping in P1 compared with a healthy control and 1 Bruton tyrosine kinase (BTK) patient (representative experiment). (F) Quantification of total mitochondrial abundance and mitochondrial activity in circulating CD19+ cells isolated from an healthy control and P1 (representative experiment). AU, arbitrary units. (G) Evaluation of pAKT, pS6, and p4EBP1 levels in B-cell bone marrow progenitors from P1 and a healthy control (2 experiments). In all graphs, **P < .01 and ***P < .001. Data are presented as means ± standard deviation. AMP, adenosine 5′-monophosphate; ATP, adenosine triphosphate; HC, healthy control; WT, wild type.
Study design
This study was approved by the Institutional Review Boards/Ethics Committees of Comitato Etico Brianza (PID-GENMET; Monza, Italy) and Baylor College of Medicine (Houston, TX). All study participants provided written informed consent.
Full methods are detailed in supplemental Materials and methods (available on the Blood Web site).
Results and discussion
Clinical phenotype
Patient 1 (P1) was born to consanguineous parents. The parents of P2 and P3 denied consanguinity (Figure 1B). Clinical manifestations started in infancy (<1 year) including severe and/or recurrent infections (detailed clinical histories in the supplementary data). Sinopulmonary infections led to bronchial wall thickening (P1), extensive bronchiectasis requiring lobectomy (P2), or calcifications (P3; supplemental Figure 1). All patients had left ventricular HCM (supplemental Figure 2; supplemental Table 1). P2 also had an interatrial communication requiring surgical correction, and P3 had severe tricuspid valve regurgitation, severe right ventricle dilatation, and pre-excitation syndrome. All patients had no imaging or laboratory signs of renal disease. For P1, neurological examination showed developmental delay associated with magnetic resonance imaging abnormalities (supplemental Figure 3). P3 had Crohn disease that required multiple bowel surgeries. All patients had absent circulating B cells and agammaglobulinemia (Table 1) requiring immunoglobulin replacement therapy. Two out of 3 patients had neutropenia, either severe (P1; neutrophil count consistently <0.5 × 109/L) or intermittent (P3), which was confirmed outside the infectious episodes and may have contributed to recurrent and severe infections.
FNIP1 variants in patients with agammaglobulinemia
Trio whole-exome sequencing was performed in all families. No candidates were identified within recognized primary immunodeficiency–associated genes.10-13 We identified distinct biallelic variants in FNIP1, which were not present in public databases (gnomAD, ESP, and 1000 Genomes) and exclusive to these families in our internal databases. For P1, the homozygous NM_133372.2:c.868C>T nonsense variant in FNIP1 is located in exon 9 and predicted to result in a premature stop codon (p.R290*). The variant was confirmed by Sanger sequencing, and each parent was a heterozygous carrier (Figure 1B; supplemental Figure 4A). A homozygous splicing donor variant (c.3306+1G>A; supplemental Figure 5) was identified in P2. Sanger sequencing confirmed the father to be a heterozygous carrier of the variant, while it was not present in the mother. Exome data were consistent with paternal uniparental disomy of chromosome 5 leading to homozygosity of this variant in P2 (supplemental Materials and methods; supplemental Figure 6). For P3, whole-exome sequencing showed a maternally inherited deletion of FNIP1 exons 9 to 18 and a paternally inherited single-nucleotide variant (c.3218delT;p.L1073Wfs*32; Figure 1B).
We evaluated functional consequences of the FNIP1 variants in the blood samples. FNIP1 messenger RNA levels were significantly decreased in P1, but not absent, most likely due to incomplete nonsense-mediated decay (Figure 1C). Because FNIP1 is expressed in activated peripheral lymphocytes,3,5 to determine whether the variants altered protein stability, we examined the presence of FNIP1 protein in stimulated cultured T cells. Immunoblotting demonstrated the complete absence of FNIP1 in all the patients (Figure 1D).
Immune-cell phenotype in FNIP1 deficiency
Analysis of the T-cell subsets showed mildly and intermittently increased CD3+ in P1 and P3. Standard lymphocyte proliferative response to specific antigens and mitogens was normal in P1 and P3 and decreased in P2 (Table 1). Using IL-2 and anti-CD28/CD3, P1 T cells showed increased apoptosis between days 7 and 11. Apoptosis was more prominent for CD8+ compared with CD4+ T cells (supplemental Figure 7). NK T cells were decreased in P1 Table 1).
Peripheral B lymphocytes were undetectable or markedly decreased in all patients Table 1). For P1, bone marrow examination displayed delayed granulocyte maturation, with no overt arrest. B-cell precursors did not show evidence of a maturation block, unlike classical agammaglobulinemia,14 although a relative increase of earlier maturation stages (pro-B and pre-B1) and a significant reduction of immature B cells were observed (Figure 1E; supplemental Figure 8).
FNIP1 deficiency is associated with altered cell metabolism
We hypothesized that human FNIP1 deficiency may hamper B-cell metabolism similar to Fnip1-deficient mice.2-8 Indeed, circulating P1 B cells exhibited increased numbers of mitochondria and mitochondrial activity relative to healthy control B cells (Figure 1F; supplemental Figure 9). Next, we examined the activation of the phosphatidylinositol 3-kinase (PI3K)/Akt pathway. We observed constitutive hyperactivation of PI3K downstream targets in P1 B-cell progenitors relative to healthy control B-cell precursors (Figure 1G). Specifically, p4EBP1 and pAkt473 were significantly activated (12.46 ± 1.86 vs 4.00 ± 0.53, P = .0002; 3.82 ± 1.78 vs 0.97 ± 0.09, P = .029). However, no difference could be found in S6 phosphorylation (1.46 ± 0.14 vs 2.70 ± 0.51, P = .14). To assess the accuracy of our approach,15 we tested NPV-BEZ 235 as a specific inhibitor of the PI3K pathway and observed that it was able to significantly decrease p4EBP1 (mean 12.46 ± 1.86 vs 2.58 ± 0.32, P = .006). These results suggest that the sensitivity to PI3K inhibition is determined by the activation of the PI3K/Akt pathway in FNIP1-deficient patients.
Mouse Fnip1 consists of 1165 amino acids and shares 91% amino acid identity with human FNIP1.5 Fnip1 plays a nonredundant role in early B-cell development and metabolism, skeletal muscle fiber type specification, and cardiac function.2-8 Our report confirms that the clinical and immunological phenotypes are strikingly overlapping (ie, HCM, pre-excitation syndrome, and early and severe B-cell defect with agammaglobulinemia). FNIP1 deficiency can be detected even in young adults. Moreover, we identified unconventional heterogeneous genetic etiologies for FNIP1 deficiency in 2 patients, expanding the variant spectrum of this novel inborn error of immunity (supplemental Figure 10A). In fact, only a limited number of cases of chromosome 5 uniparental disomy have been reported.16 Our findings argue for the clinical use of exome sequencing with copy-number variant analysis in patients with complex phenotypes. Importantly, in 1 patient, we have provided for the first time some of the functional studies (increased number of mitochondria and mitochondrial activity and 4EBP1 activation) that recapitulate the alterations described in Fnip1-deficient mice.
Some aspects nonetheless remain controversial. Contradictory results concerning the activation of the PI3K/Akt pathway have been described in Fnip1−/− mice7,17 (supplemental Table 2). Unlike Fnip1-deficient mice,17 no FNIP1 patients have had overt clinical symptoms of renal disease,9 but the presence of renal cysts has not been investigated by renal biopsies. Only 1 proband has had myopathy9 (supplemental Figure 10B). Here, we report the coexistence of heart defects other than HCM and pre-excitation syndrome in FNIP1 deficiency. Elsewhere,9 patients carrying biallelic missense variants showed milder B-cell lymphopenia, and 1 individual had only decreased immunoglobulin M (IgM) with normal IgG and elevated IgA levels. These elements suggest that FNIP1 deficiency could be a protean disorder resembling also common variable immunodeficiency phenotype. The T-cell compartment was unaffected in Fnip1−/− mice.5,7 Although patients have not shown any clinical signs of defective T-cell function, lymphocytosis and/or defective T-cell proliferation with increased apoptosis have been found in some patients (supplemental Figure 10B). Granulocytes are not affected in the Fnip1−/− mouse,7 yet 4 patients have displayed severe or intermittent neutropenia, and 2 have monocytosis. This finding is reminiscent of the neutrophil survival defect seen in Bruton tyrosine kinase–associated agammaglobulinemia.18 As in other inborn error of immunity,19,20 FNIP1 deficiency may be associated with severe or intermittent neutropenia, or even episodic normal neutrophil counts. Central nervous system involvement is present in 4 out of 6 patients. These manifestations only occurred in consanguineous pedigrees and may result either from other genetic changes not yet discovered or from FNIP1 phenotypic heterogeneity.
Collectively, we identified novel inherited FNIP1 variants causing FNIP1 deficiency, which affects B-cell survival and metabolism recapitulating the Fnip1−/− animal model. FNIP1 deficiency should be considered in patients with hypo- or agammaglobulinemia, congenital heart defects, particularly HCM, and neutropenia.
Data will be shared if requested to the corresponding author by e-mail.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank the patients and their families for participating in the study; Donna M. Muzny, Shalini N. Jhangiani, Richard A. Gibbs, and Haowei Du from the Baylor College of Medicine Human Genome Sequencing Center and Baylor-Hopkins Center for Mendelian Genomics (Houston, TX) for whole-exome sequencing and bioinformatics support; Nicholas L. Rider and Gina Cahill from the Texas Children’s Hospital William T. Shearer Center for Human Immunobiology and the Jeffrey Modell Foundation at Texas Children’s Hospital (Houston, TX) for validation studies; Francesco Canonico from the Department of Neuroradiology, University of Milan-Bicocca, San Gerardo Hospital, ASST di Monza (Monza, Italy) for the brain computed tomography study; Francesca Pluchinotta and Antonella Camporeale from Istituto di Ricovero e Cura a Carattere Scientifico Policlinico San Donato (Milan, Italy) for the cardiac magnetic resonance imaging study; Giuseppe Limoli from Unità Struttura Semplice Patologia Neonatale e Nido e Servizio di Cardiologia Pediatrica, ASST Lodi (Milan, Italy) for the cardiac ultrasound study; and GeneDx and GeneMatcher for facilitating collaboration.
This work was supported by the National Institutes of Health, National Human Genome Research Institute/National Heart, Lung, and Blood Institute (grant UM1 HG006542 to the Baylor-Hopkins Center for Mendelian Genomics), Fondecyt grant 11181222 (C.P.), Associazione Italiana Per La Ricerca Sul Cancro investigator grant 22082 (R.P.), “PG23/FROM 2017 Call for Independent Research” as part of the Rapid Assessment, Response, and Evaluation (RARE) project (to Papa Giovanni XXIII), Great Ormond Street Hospital National Institute for Health Research Biomedical Research Centre, and the Wellcome Trust (research grants 201250/Z/16/Z [E.R.] and 104807/Z14/Z [A.B.]).
Authorship
Contribution: F.S., C.P., L.D., C.B., M.I., Z.H.C.-A., and B.Y. designed and analyzed experiments; L.P.N., V.J.-C., A.B., G.F., M.Q., A.B.S., E.R., M.M., C.B., M.V., W.G.M.K., and M.M.v.O.-t.D. performed experiments; J.C.O., S.B., and S.G. provided samples and interpreted clinical and/or imaging data; J.R.L., S.v.d.M., F.B., R.P., A.B., R.B., G.C., and G.G. were involved in study design, gave intellectual input, and critically revised the manuscript; D.M. conducted immunophenotyping on bone marrow and peripheral blood mononuclear cells; and F.S., C.P, I.K.C., J.V., F.H.R., and M.S. wrote the manuscript, which was approved by all authors.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Francesco Saettini, Pediatric Hematology Department, Fondazione MBBM, University of Milano Bicocca, Via Cadore, 20900, Monza, Italy; e-mail: francescosaettini@yahoo.it.