

In this issue of Blood, identify 3 unrelated patients with clinical histories of agammaglobulinemia, recurrent infections, and hypertrophic cardiomyopathy, which are linked to loss-of-function biallelic variants in the gene encoding the metabolic regulator folliculin interacting protein-1 (FNIP1). Although studies in mice have implied the existence of metabolic checkpoints that control the development of highly metabolic immune cells, the importance of metabolic checkpoints in the prevention of primary immunodeficiency diseases (PIDs) had not been previously recognized.1
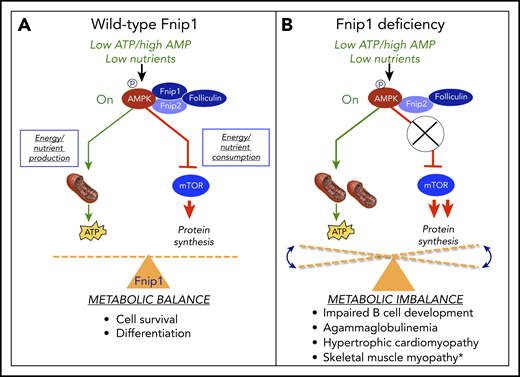
The FNIP1/FNIP2/folliculin complex helps maintain metabolic homeostasis during metabolic stress. (A) FNIP1 physically interacts with AMPK, FNIP2, and folliculin. In response to high AMP/low ATP, AMPK is phosphorylated and activated by LKB1 kinase. Activated AMPK then stimulates energy and nutrient production by increasing mitochondrial biogenesis, autophagy, and fatty acid oxidation, while inhibiting nutrient and energy consumption by mTOR. (B) FNIP1 deficiency in humans and mice results in metabolic imbalance and associated pathology. *As noted by Niehues et al.10
The FNIP1/FNIP2/folliculin complex helps maintain metabolic homeostasis during metabolic stress. (A) FNIP1 physically interacts with AMPK, FNIP2, and folliculin. In response to high AMP/low ATP, AMPK is phosphorylated and activated by LKB1 kinase. Activated AMPK then stimulates energy and nutrient production by increasing mitochondrial biogenesis, autophagy, and fatty acid oxidation, while inhibiting nutrient and energy consumption by mTOR. (B) FNIP1 deficiency in humans and mice results in metabolic imbalance and associated pathology. *As noted by Niehues et al.10
FNIP1 is an evolutionarily conserved intracytoplasmic protein originally described by Baba et al2 based on physical interactions with folliculin (a protein mutated in Birt-Hogg-Dubé syndrome), FNIP2 (a related FNIP family member), and the γ1 subunit of the master energy sensor 5′ AMP-activated protein kinase (AMPK), as reviewed by Schmidt and Linehan.3 When energy and nutrient levels are low, such as during exercise or starvation, high AMP/adenosine 5′-diphosphate ratios stimulate activation of LKB1 kinase, which phosphorylates and activates AMPK. Activated AMPK maintains metabolic homeostasis under these conditions by shutting down energy- and amino acid–consuming anabolic pathways regulated by mechanistic target of rapamycin complex 1 (mTORC1), while concurrently stimulating glucose uptake, mitochondrial biogenesis, fatty acid oxidation, and autophagy to replenish adenosine triphosphate (ATP) and nutrients required for cell growth and survival (see figure, panel A). In mice, disruption of Fnip1 either through chemical mutagenesis or gene targeting has previously been shown to result in a diverse array of biological phenotypes, including impaired B-cell development and agammaglobulinemia,4-6 disrupted invariant natural killer T (iNKT) cell development,7 hypertrophic cardiomyopathy,6 increased representation of mitochondria-rich oxidative skeletal muscle fiber types,8 and renal microcyst formation (see figure, panel B). Biochemical analyses of FNIP1-deficient murine B cells suggested either normal5,6 or increased mTORC1 activation4,9 and normal5 or increased4,6,9 AMPK activation, which were associated with increased mitochondrial number and size4,9 and increased autophagy.6,9 FNIP1-deficient murine B cells were also shown to be particularly sensitive to apoptosis in response to nutrient deprivation, and in vitro culture of FNIP1-deficient pre-B cells in nutrient-rich media partially restored B-cell development.4,9 Hence, these murine studies indicate that FNIP1 regulates the development and/or function of highly metabolic cells and tissues, including B cells, heart, skeletal muscle, and kidney, in part by maintaining metabolic balance in response to nutrient and energy availability. Of note, increased activation of both AMPK and mTOR has also been shown in mice and humans to contribute to arrhythmia syndromes and hypertrophic cardiomyopathy, potentially providing a common mechanism for the pathologies associated with FNIP1 deficiency.
Analyses of FNIP1-deficient human peripheral blood samples by Saettini et al revealed that B lymphocytes were undetectable or markedly decreased in all patients, and immunoglobulin G (IgG), IgA, IgM, and IgE levels were greatly reduced. In addition, all FNIP1-deficient patients presented with hypertrophic cardiomyopathy, consistent with phenotypes observed in FNIP1-deficient mice. However, in contrast to FNIP1-deficient mice, CD3ε+ T cells were increased and neutrophils were decreased in peripheral blood samples from 2 patients. These differences could have been caused by secondary factors, such as concurrent infections or genetic heterogeneity (which were not present in specific pathogen–free inbred murine models) or resulted from species-specific differences in signaling during development. Biochemical analyses of B cells from 1 patient showed increased phosphorylation of AKT at serine 473, indicative of mTORC2 activation, and increased phosphorylation of 4E-BP1, consistent with increased mTORC1 activation. Although AMPK activation was not specifically assessed, mitochondrial numbers and function were also increased, consistent with AMPK activation. Interestingly, a contemporary but independent study by Niehues et al10 similarly described 3 patients from 2 independent consanguineous families with severe PID, hypogammaglobulinemia, and hypertrophic cardiomyopathy resulting from autosomal recessive inherited FNIP1 deficiency. However, Niehues et al also observed muscle myopathy in all patients, characterized by enlarged abnormally shaped mitochondria in muscle biopsies, as was also noted in FNIP1-deficient mice.
Importantly, the phenotypes of FNIP1-deficient humans and mice similarly manifested in highly metabolic cell types and tissues. In addition, although constitutive (tissue-wide) targeted disruption of the BHD gene encoding folliculin in mice is lethal during embryogenesis, cell-specific disruption of folliculin was also shown to result in impaired B-cell development,5 hypertrophic cardiomyopathy, increased representation of mitochondria-rich oxidative skeletal muscle fiber types, and increased renal cyst formation and renal cancer.3 Interestingly, hematopoietic cell–specific disruption of folliculin led to a loss of hematopoietic stem cell quiescence and disrupted osteoclastogenesis, indicating essential roles for the FNIP/folliculin complex in multiple hematopoietic cell lineages. Although the phenotypes of FNIP1-deficient mice did not completely recapitulate those of FNIP1-deficient humans (eg, iNKT cells were reduced in mice but not humans), this could have been due to species-specific differences in the abilities of FNIP2 and/or folliculin to compensate for the absence of FNIP1 or differences in the balance of signaling pathways during development. These results collectively suggest that FNIP1, likely acting in a heterotrimeric complex with folliculin and FNIP2, regulates the development and survival of highly metabolic cell types, including B cells and other hematopoietic cells, in part by linking nutrient and energy availability with cell survival and differentiation.
Although variants in genes resulting in enhanced cell metabolism have long been known to contribute to hematopoietic cell cancers by helping to fuel cellular growth and proliferation, these studies by Saettini et al and Niehues et al10 are among the first to demonstrate that loss-of-function variants in genes encoding proteins involved in the regulation of cell metabolism can result in PIDs in humans. Because of the large number of proteins controlling cell metabolism, these studies further suggest that immunodeficiency diseases that also manifest in alterations in highly metabolic tissues, such as heart, skeletal muscle, and liver, may result from novel variants in genes encoding metabolic proteins. Improved understanding of the molecular mechanisms of how the FNIP/folliculin complex maintains metabolic homeostasis3 could lead to novel approaches to disrupt metabolic balance and sensitize hematopoietic cell cancers to apoptosis and/or neutralize inappropriately activated immune cells in autoimmune diseases.
Conflict-of-interest disclosure: The author declares no competing financial interests.
