Key Points
Cryo-EM structures of human factors V and Va reveal overall architecture, sites of proteolytic processing, and functional epitopes.
The structures offer molecular context for the function of factors V and Va and pioneer the analysis of coagulation factors by cryo-EM.

Abstract
Coagulation factor V (fV) is the precursor of fVa, which, together with fXa, Ca2+, and phospholipids, defines the prothrombinase complex and activates prothrombin in the penultimate step of the coagulation cascade. We solved the cryogenic electron microscopy (cryo-EM) structures of human fV and fVa at atomic (3.3 Å) and near-atomic (4.4 Å) resolution, respectively. The structure of fV reveals the entire A1-A2-B-A3-C1-C2 assembly, but with a surprisingly disordered B domain. The C1 and C2 domains provide a platform for interaction with phospholipid membranes and support the A1 and A3 domains, with the A2 domain sitting on top of them. The B domain is highly dynamic and visible only for short segments connecting to the A2 and A3 domains. The A2 domain reveals all sites of proteolytic processing by thrombin and activated protein C, a partially buried epitope for binding fXa, and fully exposed epitopes for binding activated protein C and prothrombin. Removal of the B domain and activation to fVa exposes the sites of cleavage by activated protein C at R306 and R506 and produces increased disorder in the A1-A2-A3-C1-C2 assembly, especially in the C-terminal acidic portion of the A2 domain that is responsible for prothrombin binding. Ordering of this region and full exposure of the fXa epitope emerge as necessary steps in the assembly of the prothrombin-prothrombinase complex. These structures offer molecular context for the function of fV and fVa and pioneer the analysis of coagulation factors by cryo-EM.
Introduction
Coagulation factor V (fV) was identified by Paul Owren in 1943 as a component required for the conversion of prothrombin to thrombin1,2 and was first isolated almost 4 decades later.3,4 The protein is a large (2224 residues, molecular weight [MW] 330 kDa) precursor of fVa, which, together with the enzyme fXa, Ca2+, and phospholipids defines the prothrombinase complex and rapidly converts prothrombin to thrombin in the penultimate step of the coagulation cascade.5-7 Human fV circulates in the plasma at a concentration of 20 nM and ∼20% of the total is contained in platelet α-granules where it is partially fragmented and secreted during platelet activation.8 fV is heavily glycosylated, sulfated, and phosphorylated at sites of functional significance.6 After removal of a signal peptide of 28 residues, fV secreted into plasma features the domain structure A1-A2-B-A3-C1-C2 (Figure 1), analogous to that of coagulation fVIII, with the 3 A domains homologous to ceruloplasmin. Sequence comparison shows that the A and C domains are highly conserved among human, bovine, and murine fV sequences, but the B domain is poorly conserved.9 The A1 domain (residues 1-316) is connected to the A2 domain (residues 317-709) by a short segment composed mainly of basic amino acids (residues 304-316). An acidic segment (residues 657-709) transitions the A2 domain to the large B domain (residues 710-1545), which has a main function of keeping the protein in an inactive state.7 The B domain continues into the A3 domain (residues 1546-1877), which connects to the C1 (residues 1878-2036) and C2 (residues 2037-2196) domains.

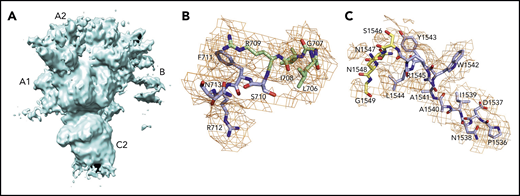
Schematic representation of the A1-A2-B-A3-C1-C2 domain organization of human fV (2196 residues) and its derivatives, fVa (1360 residues) and fVai (957 residues). The number of residues in each domain is indicated, along with relevant sites of cleavage by thrombin (R709, R1018, and R1545) and APC (R306 and R506). Removal of the large B domain (836 residues) by thrombin produces the fVa heterodimer composed of heavy (A1-A2) and light (A3-C1-C2) chains containing 709 and 651 residues, respectively. Further cleavage by APC at R306 and R506 produces fVai. The X-ray structure of bovine fVai was reported previously.31 This structure contains no information on the sites of cleavage in the A2 and B domains and also carries disorder in the A1 domain around R306. The cryo-EM structures of human fV and fVa are reported in this study (Figure 2). The structure of fV contains all residues of the A1-A2-A3-C1-C2 assembly and 14 additional residues in the B domain.
Schematic representation of the A1-A2-B-A3-C1-C2 domain organization of human fV (2196 residues) and its derivatives, fVa (1360 residues) and fVai (957 residues). The number of residues in each domain is indicated, along with relevant sites of cleavage by thrombin (R709, R1018, and R1545) and APC (R306 and R506). Removal of the large B domain (836 residues) by thrombin produces the fVa heterodimer composed of heavy (A1-A2) and light (A3-C1-C2) chains containing 709 and 651 residues, respectively. Further cleavage by APC at R306 and R506 produces fVai. The X-ray structure of bovine fVai was reported previously.31 This structure contains no information on the sites of cleavage in the A2 and B domains and also carries disorder in the A1 domain around R306. The cryo-EM structures of human fV and fVa are reported in this study (Figure 2). The structure of fV contains all residues of the A1-A2-A3-C1-C2 assembly and 14 additional residues in the B domain.
Conversion of fV to fVa is catalyzed by thrombin upon cleavage at R709, R1018, and R1545 and release of the entire B domain in 2 fragments (Figure 1). The sites of cleavage by thrombin are conserved across mammalian species.10 The large B domain maintains the inactive state of fV,7,11,12 and its deletions produce derivatives functionally equivalent to fVa.12,13 Removal of a basic region of the B domain (residues 963-1008) by proteolysis12 or alternative splicing14 yields derivatives that are constitutively active in the prothrombinase complex and unmasks a binding site (residues 1493-1537) for tissue factor pathway inhibitor-α (TFPI-α), which blocks procoagulant function12 and produces a mild bleeding phenotype.14 Removal of the entire B domain splits the protein into the A1-A2 heavy chain (MW 105 kDa) and A3-C1-C2 light chain (MW 74 kDa), which assemble as a noncovalent Ca2+-linked heterodimer. This form of fVa is extremely stable, and its partial inactivation is produced proteolytically by cleavage at R506 in the A2 domain by activated protein C (APC), with the assistance of protein S. Complete inactivation is achieved by 2 additional cleavages at R306 in the A1 domain and R679 in the C terminus of the A2 domain. Cleavage of fV at R506 by APC produces an alternative form of fV that assists APC inactivation of fVIIIa with the assistance of protein S.7,15,16 Hence, fV features a procoagulant role as a precursor of fVa to amplify the coagulation response and an anticoagulant role as an assistant of APC in the feedback regulation of the same response. Interestingly, fVa also functions as a cofactor of fXI activation by thrombin.17 The regulation of thrombin generation is critically dependent on the levels of fVa, and failure to control fVa activity may result in either bleeding or thrombotic complications. Complete deficiency of fV in mice results in midembryogenic lethality or fatal perinatal hemorrhage.18 In contrast, the bleeding manifestation of severe fV deficiency in human patients varies dramatically.7 The physiological importance of the downregulation of fVa activity by APC is demonstrated by the variant R506Q, known as fVLeiden, which is a common genetic risk factor for venous thrombosis in humans.19,20 Mice carrying the homologous mutation exhibit normal survival, but demonstrate a marked increase in spontaneous tissue fibrin deposition compared with wild-type mice.21 The naturally occurring fVLeiden also impairs the anticoagulant properties of fV.22,23
Although the importance of fV and fVa in blood coagulation is well established, a molecular understanding of their function remains incomplete. Previous electron microscopy images of fVa have revealed globular molecules with several smaller appendicular structures but lacking the long tails seen in fV and concluded that the A and C domains have a globular shape with the connecting B domain contained in a 2-stranded tail that is released by thrombin cleavage.24 Other electron microscopy studies of fVa interacting with lipid surfaces containing phosphatidylserine revealed 2 distinct domains of the protein of different size.25 Atomic force microscopy topographic images have shown that fVa is somewhat dynamic in the C1 and C2 domains.26 Several structural models of fVa have been proposed.27-30 A pioneering X-ray structure of bovine fVa inactivated by APC (fVai) lacks the A2 domain (Figure 1) and shows the C1 and C2 domains defining a platform that interacts with phospholipid membranes and supports the A1 and A3 domains.31 However, absence of the A2 domain in this structure offers no clues about the sites of cleavage by APC and thrombin or about the architecture of epitopes responsible for interaction with fXa, prothrombin, and APC. The lack of structural information on fV precludes comparison with fVa and assessment of the role of the B domain. We report cryogenic electron microscopy (cryo-EM) structures of human fV and fVa that fill existing gaps in our understanding of the architecture of these key coagulation proteins. The results represent a promising start for future structural analysis of coagulation complexes by cryo-EM.
Methods
Materials
Human fVa (F0931, 50 μg) and human fV (F0681, 50 μg) were purchased from Sigma-Aldrich (St Louis, MO). Human fV was further purified by size exclusion chromatography on a Super-dex 10/300 Mono Increase Column in 20 mM N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid, 150 mM NaCl, and 5 mM CaCl2, at a flow rate of 0.5 mL/min. SEC purification of human fVa yielded the separate heavy (A1-A2 domains) and light (A3-C1-C2 domains) chains, but failed to produce a reconstituted heterodimer stable enough for structure determination. Therefore, fVa purchased from 3 different vendors was assessed for purity by silver stain gels and used without further purification. Both fVa and fV (supplemental Figure 1, available on the Blood Web site) were diluted to a concentration of 0.1 mg/mL in 20 mM N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid, 150 mM NaCl, and 5 mM CaCl2 for vitrification. The cryo-EM grids used were Quantifoil 2/2 holey carbon grids, glow discharged before vitrification, which was performed on a Vitrobot Mark IV (FEI). The grids were blotted for 2 seconds at −1 blot force with no humidity control. Data were collected using EPU (Thermo Fisher Scientific) on a Titan Krios G3 cryo–transmission electron microscope, operating at 300 kV and equipped with a GIF BioQuantum 968 energy filter and a Gatan K2 Summit (Gatan) Direct Electron Detector, with a pixel size of 1.1 Å, a dose of 1.65 e−/Å2, and a defocus value of between −1 and −2.5 μm.
Image processing, map calculation, and model building
Image stacks were aligned, binned, and dose weighted with MotionCor2.32 Contrast transfer function (CTF) determination was performed with CTFFind4.33 Motion-corrected and CTF-estimated images were curated according to CTF estimations of a maximum resolution of 4 Å. For fV, a 2 631 626 particles were initially selected, using the Relion34 Laplacian-of-Gaussian method with a minimum diameter of 60 Å and a maximum diameter of 160 Å and extracted into a box size of 256 Å. Several cycles of 2-dimensional (2D) classification, as implemented in Relion were run to remove low-quality particle images and yield templates for template picking, as implemented in Relion. The resulting particles were imported into cryoSPARC,35 and several more rounds of 2D classification were used to further improve the quality of the data set. The remaining particles were used to generate an initial map for 3D classification and subsequent refinement. After a final CTF refinement of 299 182 particles, a map of 4.1 Å was reached that showed low-resolution density, most likely corresponding to the disordered B domain. A mask was generated to remove the low-resolution density, and further local refinement yielded a map of 3.3 Å. For fVa, 699 611 particles were initially selected by the Relion Laplacian-of-Gaussian method, with a minimum diameter of 60 Å and a maximum diameter of 140 Å, and extracted into a box size of 256 Å. As with fV, several cycles of 2D classification were performed, but the resultant particles were subjected to additional rounds of 3D classification in Relion to remove low-quality and broken particles consisting of separate heavy and light chains. Cleaned particles were exported to cryoSPARC for further 2D classification and generation of an initial model. After 3D classification and CTF refinement, a final set of 63 745 particles yielded a map of 4.7 Å. No distinct low-resolution density was apparent. A final masked local refinement generated a map of 4.4 Å. Model building proceeded, using the coordinates 1SDD of bovine fVai31 and 4BXS of fV from the snake Pseudonaja textilis30 as templates to generate models for the A1, A2, A3, C1, and C2 domains with Modeler.36 Initial models were docked onto map density by the fit-in-map procedure of Chimera, before being subjected to multiple iterative rounds of real-space refinement in Phenix37 and manual model-building in COOT.38 Relevant parameters of the cryo-EM structures of fV and fVa are summarized in Table 1. The solvent exposure of individual residues was calculated as reported elsewhere.39 Representative 2D class averages and gold-standard Fourier shell correlation of masked refinement of the fV and fVa maps are reported in supplemental Figures 2A-B and 3A-B.
Results
The current X-ray structure of bovine fVai (protein database [PDB] ID, 1SDD) contains the A1-A3-C1-C2 domains (Figure 1) bearing disorder in several regions, including the site of cleavage by APC at R306 in the A1 domain.31 Neither the sites of cleavage by thrombin or APC, nor the epitopes for fXa, APC, and prothrombin binding could be located in this structure, thereby leaving major questions about fV and fVa function unanswered. Nonetheless, the importance of this structure is that it reveals the C domains aligned edge to edge, forming a platform involved in membrane binding and upon which the A domains rest side by side, around a pseudo-3-fold axis similar to that observed in ceruloplasmin. The cryo-EM structure of fV, solved at atomic (3.3 Å) resolution, reveals the entire A1-A2-A3-C1-C2 domains (1360 residues) for the first time, with the A1-A3-C1-C2 domains arranged as seen in the bovine fVai structure (root mean square deviation [RMSD] = 1.29 Å over 765 Cα atoms) and the A2 domain wedged between the A1 and A3 domains, with no contacts with the C domains (Figure 2). This portion of the structure is also similar to that of fV from the snake P textilis30 (PDB ID: 4BXS; RMSD = 1.80 Å over 972 Cα atoms for fV). The large B domain containing 836 residues is surprisingly dynamic and almost fully disordered, except for the N-terminal 710SFRN713 segment connecting to the A2 domain and the C-terminal 1536PDNIAAWYLR1545 segment connecting to the A3 domain (Figure 3). Unfortunately, these short segments do not visualize the basic (residues 963-1008) and acidic (residues 1493-1537) regions that are responsible for constitutive activation and binding of TFPIα.7,12,13,40 The sites of thrombin activation of fV at R709 and R1545 are clearly visible in the A2 and B domains and exposed to solvent for proteolytic attack (Figure 2B-C), but the sites of APC cleavage at R306 and R506 are 75% buried (Figure 4A-B). Putative epitopes for fXa, APC, and prothrombin are fully resolved. The structure of fVa solved at nearly atomic (4.4 Å) resolution reveals the organization of the A1-A2-A3-C1-C2 domains, but is considerably more disordered than that of fV, with fewer (1181 of 1360) resolved residues, especially in the A2 (294 of 393) and A3 (295 of 332) domains. Yet, the structure overlaps well with those of fV (RMSD = 2.26 Å over 972 Cα atoms), bovine fVai (RMSD = 2.47 Å over 676 Cα atoms), and fV from the snake P. textilis30 (RMSD = 2.50 Å over 1062 Cα atoms). Increased dynamics in the structure of fVa may be the result of activation and release of the entire B domain, with formation of a 2-chain, noncovalently linked heterodimer intrinsically less stable than its 1-chain precursor. The disorder in fVa affects mainly the C2 domain and the epitope of prothrombin binding located toward the C terminus of the A2 domain. The ordering of this flexible region may have functional consequences in the assembly of prothrombinase. In contrast, the sites of APC cleavage at R306 and R506 leading to fVa inactivation are 60% exposed to solvent, unlike that in fV where density from a disordered B domain blocks the accessibility of those residues (Figure 4C-D).
![Cryo-EM structures. fV (A-C) and fVa (D-F) structures are shown. The 2 proteins are rendered in surface representation with the constitutive domains colored in wheat [A1], pale green [A2], light blue [B], pale yellow [A3], light pink [C1], and pale cyan [C2]. The structure of fV (A-C) was solved at 3.3 Å resolution and features the C domains aligned edge to edge, forming a platform involved in membrane binding and upon which the A domains rest side by side, arranged around a pseudo-3-fold axis. The A1-A2-A3-C1-C2 domain assembly is resolved in its entirety. The sites of thrombin activation at R709 and R1545 (magenta) are clearly visible in the A2 and B domains and are exposed to solvent for proteolytic attack. The sites of APC cleavage at R306 and R506 (red) are 75% buried. The exact orientation of the side chains at the sites of cleavage by thrombin and APC were assigned with sufficient confidence in the context of the resolution obtained (shown also in Figures 3B-C and 4). The structure also reveals the spikes in the C domain responsible for membrane docking (olive) and the epitopes of binding for fXa (orange), APC (blue), and prothrombin (gray), which occupies separate segments in the C-terminal of the A2 domain and the C2 domain (arrows labeled proT). Most of the fXa epitope is buried under the short segment 373DESF376 (asterisk) of the A2 domain in both fV (C) and fVa (F). A rearrangement of this segment appears necessary to fully expose the epitope upon assembly of prothrombinase. The B domain is very dynamic; only 14 residues are resolved in the connection to the A2 domain (710SFRN713 segment) and the A3 domain (1536PDNIAAWYLR1545 segment). The structure of fVa (D-F) was solved at 4.4 Å resolution and is more disordered than that of fV, with fewer (1181 of 1360) residues resolved in the A1 (294 of 316), A2 (294 of 393), A3 (295 of 332), and C2 (139 of 160) domains. Overall, the arrangement of the A1-A2-A3-C1-C2 domain assembly is similar to that of fVa (RMSD = 2.26 over 972 Cα atoms). The disorder in fVa affects the spikes in the C2 domain (D) and the entire epitope of prothrombin binding (gray) that are missing in the density map (E-F). Structuring of the prothrombin epitope may be necessary to assemble a functional prothrombinase-prothrombin complex. Importantly, the sites of APC cleavage at R306 and R506 leading to fVa inactivation are 60% exposed to solvent (E-F), unlike what is seen in fV (B-C). The structures also provide context for the functional consequences of posttranslational modifications and mutations in the C1 and C2 domains that reduce binding of fV to phospholipid membranes and perturb its cofactor activity. The site of glycosylation at N2181 and residue A2086 are clearly visible on the surface of the C2 domain, but residue W1920 is buried under Y1903 in the C1 domain (A,D).](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/137/22/10.1182_blood.2021010684/1/m_bloodbld2021010684f2.png?Expires=1769316357&Signature=OVzwdJHqWdO7eXWHqA-ciJTxbQf~A-Qsb7G~9KtdJN2GNklwt8uTci-oKzKfDm4b5DKtPnsEqUn~CvCSCWModmtkg0FS92QVK3DOZFTPUdj3fWbbsJXd2jEjBZMveLfYFPAP-wtdadpi2ePJ~ymHP4iY7nB4KNjGi0S6vUgrx8XG4aOPfC~7QztIwwXIEa75vgqqPAcpQOylqftTn5X1YQOyNv7rBlRxB17pFZZ04pNVqpTSIV~IljNPSYNsIu0e-QGI~xyveYN9g-KaIs5gBWfxzcDJ1YzLKOGqo~l6SqbF5Sd0P-ShBU8xq5h97Jiqiby-6lcZkSSPwvij~NBlgw__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Cryo-EM structures. fV (A-C) and fVa (D-F) structures are shown. The 2 proteins are rendered in surface representation with the constitutive domains colored in wheat [A1], pale green [A2], light blue [B], pale yellow [A3], light pink [C1], and pale cyan [C2]. The structure of fV (A-C) was solved at 3.3 Å resolution and features the C domains aligned edge to edge, forming a platform involved in membrane binding and upon which the A domains rest side by side, arranged around a pseudo-3-fold axis. The A1-A2-A3-C1-C2 domain assembly is resolved in its entirety. The sites of thrombin activation at R709 and R1545 (magenta) are clearly visible in the A2 and B domains and are exposed to solvent for proteolytic attack. The sites of APC cleavage at R306 and R506 (red) are 75% buried. The exact orientation of the side chains at the sites of cleavage by thrombin and APC were assigned with sufficient confidence in the context of the resolution obtained (shown also in Figures 3B-C and 4). The structure also reveals the spikes in the C domain responsible for membrane docking (olive) and the epitopes of binding for fXa (orange), APC (blue), and prothrombin (gray), which occupies separate segments in the C-terminal of the A2 domain and the C2 domain (arrows labeled proT). Most of the fXa epitope is buried under the short segment 373DESF376 (asterisk) of the A2 domain in both fV (C) and fVa (F). A rearrangement of this segment appears necessary to fully expose the epitope upon assembly of prothrombinase. The B domain is very dynamic; only 14 residues are resolved in the connection to the A2 domain (710SFRN713 segment) and the A3 domain (1536PDNIAAWYLR1545 segment). The structure of fVa (D-F) was solved at 4.4 Å resolution and is more disordered than that of fV, with fewer (1181 of 1360) residues resolved in the A1 (294 of 316), A2 (294 of 393), A3 (295 of 332), and C2 (139 of 160) domains. Overall, the arrangement of the A1-A2-A3-C1-C2 domain assembly is similar to that of fVa (RMSD = 2.26 over 972 Cα atoms). The disorder in fVa affects the spikes in the C2 domain (D) and the entire epitope of prothrombin binding (gray) that are missing in the density map (E-F). Structuring of the prothrombin epitope may be necessary to assemble a functional prothrombinase-prothrombin complex. Importantly, the sites of APC cleavage at R306 and R506 leading to fVa inactivation are 60% exposed to solvent (E-F), unlike what is seen in fV (B-C). The structures also provide context for the functional consequences of posttranslational modifications and mutations in the C1 and C2 domains that reduce binding of fV to phospholipid membranes and perturb its cofactor activity. The site of glycosylation at N2181 and residue A2086 are clearly visible on the surface of the C2 domain, but residue W1920 is buried under Y1903 in the C1 domain (A,D).
Cryo-EM structures. fV (A-C) and fVa (D-F) structures are shown. The 2 proteins are rendered in surface representation with the constitutive domains colored in wheat [A1], pale green [A2], light blue [B], pale yellow [A3], light pink [C1], and pale cyan [C2]. The structure of fV (A-C) was solved at 3.3 Å resolution and features the C domains aligned edge to edge, forming a platform involved in membrane binding and upon which the A domains rest side by side, arranged around a pseudo-3-fold axis. The A1-A2-A3-C1-C2 domain assembly is resolved in its entirety. The sites of thrombin activation at R709 and R1545 (magenta) are clearly visible in the A2 and B domains and are exposed to solvent for proteolytic attack. The sites of APC cleavage at R306 and R506 (red) are 75% buried. The exact orientation of the side chains at the sites of cleavage by thrombin and APC were assigned with sufficient confidence in the context of the resolution obtained (shown also in Figures 3B-C and 4). The structure also reveals the spikes in the C domain responsible for membrane docking (olive) and the epitopes of binding for fXa (orange), APC (blue), and prothrombin (gray), which occupies separate segments in the C-terminal of the A2 domain and the C2 domain (arrows labeled proT). Most of the fXa epitope is buried under the short segment 373DESF376 (asterisk) of the A2 domain in both fV (C) and fVa (F). A rearrangement of this segment appears necessary to fully expose the epitope upon assembly of prothrombinase. The B domain is very dynamic; only 14 residues are resolved in the connection to the A2 domain (710SFRN713 segment) and the A3 domain (1536PDNIAAWYLR1545 segment). The structure of fVa (D-F) was solved at 4.4 Å resolution and is more disordered than that of fV, with fewer (1181 of 1360) residues resolved in the A1 (294 of 316), A2 (294 of 393), A3 (295 of 332), and C2 (139 of 160) domains. Overall, the arrangement of the A1-A2-A3-C1-C2 domain assembly is similar to that of fVa (RMSD = 2.26 over 972 Cα atoms). The disorder in fVa affects the spikes in the C2 domain (D) and the entire epitope of prothrombin binding (gray) that are missing in the density map (E-F). Structuring of the prothrombin epitope may be necessary to assemble a functional prothrombinase-prothrombin complex. Importantly, the sites of APC cleavage at R306 and R506 leading to fVa inactivation are 60% exposed to solvent (E-F), unlike what is seen in fV (B-C). The structures also provide context for the functional consequences of posttranslational modifications and mutations in the C1 and C2 domains that reduce binding of fV to phospholipid membranes and perturb its cofactor activity. The site of glycosylation at N2181 and residue A2086 are clearly visible on the surface of the C2 domain, but residue W1920 is buried under Y1903 in the C1 domain (A,D).

Disorder in the B domain limits resolution of the architecture of this portion of fV. (A) Several regions of the domain are detected as streaks that cover most of the A2 and A3 domains, as shown by the unmasked map at 4.1 Å resolution. The orientation of fV corresponds to that in Figure 2B, after rotation of 90° around the y-axis. The only ordered regions of the B domain (836 residues) are limited to 14 residues connecting to the A2 and A3 domains. (B) The N-terminal 710SFRN713 segment (light blue) connects to the A2 domain (pale green) (B) and the C-terminal 1536PDNIAAWYLR1545 segment (light blue) connects to the A3 domain (pale yellow) (C).
Disorder in the B domain limits resolution of the architecture of this portion of fV. (A) Several regions of the domain are detected as streaks that cover most of the A2 and A3 domains, as shown by the unmasked map at 4.1 Å resolution. The orientation of fV corresponds to that in Figure 2B, after rotation of 90° around the y-axis. The only ordered regions of the B domain (836 residues) are limited to 14 residues connecting to the A2 and A3 domains. (B) The N-terminal 710SFRN713 segment (light blue) connects to the A2 domain (pale green) (B) and the C-terminal 1536PDNIAAWYLR1545 segment (light blue) connects to the A3 domain (pale yellow) (C).
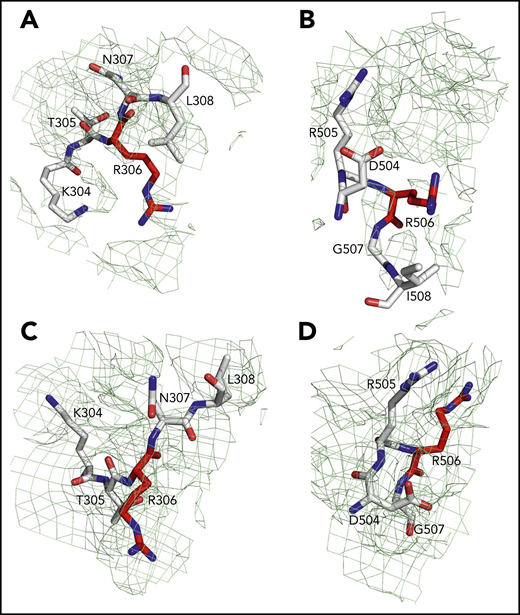
Sites of cleavage by APC in fV and fVa. Residues R306 (A,C) and R506 (B,D) are 75% buried in fV (A-B) but 60% exposed in fVa (C-D). Mesh refers to residues 304 to 308 (A,C), 504 to 508 (B), and 504 to 507 (D) and was contoured at 1 σ.
Sites of cleavage by APC in fV and fVa. Residues R306 (A,C) and R506 (B,D) are 75% buried in fV (A-B) but 60% exposed in fVa (C-D). Mesh refers to residues 304 to 308 (A,C), 504 to 508 (B), and 504 to 507 (D) and was contoured at 1 σ.
The similar organization of fV and fVa lends confidence to using the structure of fV as a good template for all functionally important regions of fVa. The 2 C domains in the cryo-EM structure of fV are similar to each other (RMSD = 1.19 Å over 120 Cα atoms) and are shaped as distorted jelly-roll β barrels, as shown in the X-ray structures of bovine fVai31 and the recombinant C2 domain of human fVa and fVIIIa.41,42 Together, they define a platform for interaction with the membrane on 1 side and support for the A1-A2-A3 domains on the other side, to yield a structure that fits a box 120 Å high, 80 Å across, and 50 Å deep (Figure 2B), consistent with previous EM measurements.24 After activation to fVa, the 2 C domains become less similar to each other (RMSD = 3.57 Å over 127 Cα atoms), with most of the difference residing in the C2 domain and specifically in the β-hairpin loops or spikes at the bottom of the C1-C2 platform supposed to dock fVa on the phospholipid surface. The docking platform in the C1 domain of fV contains residues Y1903, W1904, Y1917, L1957, and especially Y1956 (Figure 2A-B), which constitutes a major determinant of interaction with the membrane.43-45 The analogous docking platform of the C2 domain involves residues W2063, W2064, and L2116. In the X-ray structure of the recombinant C2 domain of human fV41 (PDB ID, 1CZT), W2063 and W2064 are considered primary determinants for insertion into the membrane bilayer,46 together with electrostatic interactions.47 They are trapped into the A3 domain from a neighboring molecule in the crystal of bovine fVai,31 but are exposed to solvent in the cryo-EM structure. The spikes in the C1 domain of fVa are organized as in fV, but only L2116 is visible in the C2 domain because of increased flexibility of this region in fVa (Figure 2D-E). These observations support a functional equivalence for the C1 domain between fV and fVa,44 but not for the C2 domain, because of the different orientation of W2063 and W2064.46
Of the 3 A domains, A3 is slightly more similar to A1 (RMSD = 1.22 Å over 226 Cα atoms) than A2 (RMSD = 2.27 Å over 222 Cα atoms). The A1 domain also changes the least when transitioning to fVa (RMSD = 1.64 Å over 264 Cα atoms) and the A2 domain changes the most (RMSD = 2.26 Å over 256 Cα atoms). Each A domain contains 2 linked cupredoxin-like β-barrels and shares similarity with the A domains of ceruloplasmin. The A1-A2 domains interact with the A3-C1-C2 domains through a surface area of 5755 Å2 that is reduced slightly to 5546 Å2 between the heavy and light chains of fVa. The sites of cleavage by APC at R506 and R306 are visible in the density map for both fV and fVa (Figures 2 and 4). The Cα-Cα distance between the 2 residues is ∼35 Å, supporting independent cleavage at both sites. Residue R506 sits ∼100 Å from the bottom of the plane defined by the C1 and C2 domains, in agreement with the distance calculated for the active site of APC above the membrane from FRET measurements.48 Solvent exposure is 60% for both residues in fVa, challenging the conclusion from molecular modeling29 and mutagenesis studies49 that prior cleavage at R506 is required for exposure of R306. Interestingly, R506 and R306 are 75% buried in fV, implying that conversion to fVa primes these residues for proteolytic attack by APC. The molecular basis of this difference may be linked to the dynamics of the C-terminal segment of the A2 domain. In fV, the segment folds back toward R306 in the A1 domain (Figure 3B), but in fVa it becomes disordered from V654 through R709 at the C terminus of the heavy chain. Overall, the A2 domain of fVa is more dynamic but similar to that of fV (RMSD = 2.26 Å over 256 Cα atoms), with the core organized in more extensive β-sheets in fV. Importantly, R709 is fully exposed to solvent in fV and shows its connection to the short N-terminal segment of the B domain that is visible in the density map (Figure 3B). The bulk of the B domain presumably loops around the protein in a highly dynamic conformation (Figure 3A) before connecting to the other thrombin cleavage site at R1545 on the opposite side of the molecule (Figure 3C). We note that, although the exact orientation of individual side chains should always be considered in the context of the resolution obtained, it could be assigned with sufficient confidence for the sites of cleavage by thrombin and APC (Figures 3 and 4).
The cryo-EM structures of fV and fVa also reveal putative binding epitopes. Binding of fV to fXa is weak and produces only a modest enhancement of prothrombin activation. Limited proteolysis of the region of the B domain around R1545 promotes high-affinity binding to fXa.50 Assembly of fVa within prothrombinase produces a drastic (>10 000-fold) increase of the rate of prothrombin conversion to thrombin and directs prothrombin activation along the meizothrombin pathway, whereas, in the absence of fVa, prothrombin activation is considerably slower and proceeds along the alternative prethrombin-2 pathway.51,52 The region 323 to 331 of the A2 domain of fVa has been proposed as an epitope for fXa binding.53,54 Most of this region is buried under the short segment 373DESF376 of the A2 domain in both fV and fVa (Figure 2C,F, asterisks). A rearrangement of this segment appears necessary to fully expose the epitope. The nearby region 311 to 325 at the boundary between the A1 and A2 domains is a potential epitope for APC recognition55 and is exposed to solvent in both fV and fVa (Figure 2C,F). Other studies have identified a cluster of amino acids located on the C-terminal portion of the heavy chain (residues 680-709) in the A2 domain as epitopes involved in prothrombin activation and prothrombinase function.56,57 A model of the prothrombinase-prothrombin complex supports the C terminus of the A2 domain as the locale for fXa and prothrombin docking.28 The segment 659DDDED663 controls the rate of cleavage of prothrombin by prothrombinase,58 and segment 695DYDYQNRLAAALGIR709 controls prothrombin binding.40,47 The sequence 695DYDYQ699 functions as a competitive inhibitor of prothrombin activation by prothrombinase and shifts the pathway of activation from meizothrombin to prethrombin-2.59 These regions are completely disordered in the structure of fVa, but appear as exposed patches 37 Å apart in the structure of fV (Figure 2C, arrows indicating gray surfaces). The segment 655 to 709 of fV (Figure 3B) may become highly dynamic in fVa and relinquish its interaction with the rest of the A2 domain, as seen in fV, to function as a “tentacle” protruding into the solvent. Ordering of this tentacle and full exposure of the fXa epitope by removal of the covering segment 373DESF376 may be necessary to assemble a functional prothrombinase-prothrombin complex. However, we stress that most of the current experimental evidence on the epitopes of prothrombin and fXa is indirect. Conclusive assignments must await a cryo-EM structure of the prothrombin-prothrombinase complex.
The position of the metal binding sites for Ca2+ and Cu2+, first reported in the structure of bovine fVai,31 could not be confirmed in the cryo-EM structures of fV and fVa because of the lack of clear density for the metal ions. Ca2+ binds to the A1 domain and is coordinated by the side chains of D11 and D112 and the backbone O atoms of K93 and D108. A proper arrangement for these residues is observed in the structure of fV and fVa, but no density can be assigned to the cation. Similarly, H1802, H1804, and D1844 in the A3 domain of bovine fVai coordinate Cu2+ and appear in this same orientation, favoring Cu2+ binding in the structures of fV and fVa, but with no density associated with the metal.
Discussion
The results of our work fill existing gaps in our molecular understanding of fV and fVa. Especially important is the new information on the A2 domain and the spatial organization of the epitopes of binding of fXa, APC, and prothrombin. In addition, the sites of thrombin activation at R709 and R1545 and the sites of APC cleavage at R306 and R506 are fully resolved. Residue R506 associated with fVLeiden15,16,19,21,22,60 is exposed to solvent for proteolytic attack by APC in fVa but not in fV and sits atop the A2 domain (Figure 2C,F), in close proximity to the epitope that recognizes APC. Hence, proteolytic attack at R506 by APC, leading to fVa inactivation, most likely takes place as a rapid “rigid-body” association limited only by diffusion61 and at a rate significantly faster than in fV, in agreement with biochemical data.49 The isosteric R506Q substitution does not affect exposure of the side chain but strongly reduces electrostatic coupling with the primary specificity pocket of APC, thereby compromising the kcat/Km for inactivation of fVa. Cleavage at R306 is also required for fVa inactivation by APC.6,7 This residue is exposed to solvent in fVa, as well, but not in fV, suggesting that activation of fV produces conformational changes that prime R306 and R506 for proteolysis. The structure of fVa suggests independent cleavage at the 2 separate sites, rather than an ordered sequence of cleavage at R506 first, followed by R306, as suggested by recent molecular modeling29 and mutagenesis studies.49 The site of thrombin activation at R1545 is visible in the short fragment that connects the highly dynamic B domain to the A3 domain and is exposed to solvent. About 67 Å away from it, residue R709 in the A2 domain is fully exposed to solvent and is positioned in close proximity to the site of APC cleavage at R306. Thrombin attack at these sites is not sterically impeded and should be limited only by diffusion.
The cryo-EM structures also provide context for the functional consequences of posttranslational modifications and mutations in the C1 and C2 domains that reduce binding of fV to phospholipid membranes and perturb its cofactor activity. Perturbation of the spikes is likely the molecular origin of the effects. Residue N2181 is close to W2063 and W2064 and is exposed to solvent (Figure 2A,D). Glycosylation of this residue in the fV1 isoform, accounting for 33% of the circulating fV,62 may easily cause steric hindrance for binding to phospholipid membranes. Local electrostatic repulsion and reduced binding to phospholipid membranes may explain the functional defect reported recently for fVBesançon (A2086D)63 affecting the nearby, solvent-exposed A2086 (Figure 2A,D). On the other hand, the mutation W1920R in fVNara64 affects a residue completely buried under Y1903 and W1904 in the C1 domain (Figure 2A,D) in a hydrophobic cage lined by L1901, P1933, I1935, P2017, and L2026. Introduction of Arg in this hydrophobic cage most likely disrupts the orientation of the spikes above it.
The B domain of fV is surprisingly dynamic, but it provides the connectivity necessary to order the A1-A2-A3-C1-C2 assembly and to reveal its entire architecture for the first time. Removal of the B domain during transition to fVa increases disorder of the A1-A2-A3-C1-C2 assembly, which likely influences the conformation of prothrombinase. This effect makes a cryo-EM structure of the prothrombin-prothrombinase complex of paramount importance in establishing the conformational plasticity of its components and in conclusively assigning the epitopes of interaction between fVa, prothrombin, and fXa. The structures reported herein will facilitate the task.
Structures have been deposited in the Protein Data Bank (accession codes 7KVE for fV; 7KXY for fVa). Protein structures have been released to the public, and are also available by e-mail request to the corresponding author (enrico@slu.edu).
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank Tracey Baird for her assistance with the illustrations.
This study was supported, in part, by National Institutes of Health (NIH), National Heart, Lung, and Blood Institute grants HL049413, HL139554, and HL147821 (E.D.C.). M.J.R. and J.A.J.F. are supported by the Washington University Center for Cellular Imaging, which is funded, in part, by Washington University School of Medicine, the Children’s Discovery Institute of Washington University, and St Louis Children’s Hospital (grants CDI-CORE-2015-505 and CDI-CORE-2019-813); the Foundation for Barnes-Jewish Hospital (grant 3770); the Washington University Diabetes Research Center (NIH, National Institute of Diabetes and Digestive and Kidney Diseases grant DK020579); and The Alvin J. Siteman Cancer Center at Barnes-Jewish Hospital and Washington University School of Medicine (NIH, National Cancer Institute grant CA091842).
Authorship
Contribution: E.A.R. performed biochemical preparations, structure determination, and analysis; M.J.R. and J.A.J.F. performed cryo-EM data acquisition; and all authors analyzed the results and prepared the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Enrico Di Cera, Edward A. Doisy Department of Biochemistry and Molecular Biology, Saint Louis University School of Medicine, 1100 S Grand Blvd, St Louis, MO 63104; e-mail: enrico@slu.edu.



![Cryo-EM structures. fV (A-C) and fVa (D-F) structures are shown. The 2 proteins are rendered in surface representation with the constitutive domains colored in wheat [A1], pale green [A2], light blue [B], pale yellow [A3], light pink [C1], and pale cyan [C2]. The structure of fV (A-C) was solved at 3.3 Å resolution and features the C domains aligned edge to edge, forming a platform involved in membrane binding and upon which the A domains rest side by side, arranged around a pseudo-3-fold axis. The A1-A2-A3-C1-C2 domain assembly is resolved in its entirety. The sites of thrombin activation at R709 and R1545 (magenta) are clearly visible in the A2 and B domains and are exposed to solvent for proteolytic attack. The sites of APC cleavage at R306 and R506 (red) are 75% buried. The exact orientation of the side chains at the sites of cleavage by thrombin and APC were assigned with sufficient confidence in the context of the resolution obtained (shown also in Figures 3B-C and 4). The structure also reveals the spikes in the C domain responsible for membrane docking (olive) and the epitopes of binding for fXa (orange), APC (blue), and prothrombin (gray), which occupies separate segments in the C-terminal of the A2 domain and the C2 domain (arrows labeled proT). Most of the fXa epitope is buried under the short segment 373DESF376 (asterisk) of the A2 domain in both fV (C) and fVa (F). A rearrangement of this segment appears necessary to fully expose the epitope upon assembly of prothrombinase. The B domain is very dynamic; only 14 residues are resolved in the connection to the A2 domain (710SFRN713 segment) and the A3 domain (1536PDNIAAWYLR1545 segment). The structure of fVa (D-F) was solved at 4.4 Å resolution and is more disordered than that of fV, with fewer (1181 of 1360) residues resolved in the A1 (294 of 316), A2 (294 of 393), A3 (295 of 332), and C2 (139 of 160) domains. Overall, the arrangement of the A1-A2-A3-C1-C2 domain assembly is similar to that of fVa (RMSD = 2.26 over 972 Cα atoms). The disorder in fVa affects the spikes in the C2 domain (D) and the entire epitope of prothrombin binding (gray) that are missing in the density map (E-F). Structuring of the prothrombin epitope may be necessary to assemble a functional prothrombinase-prothrombin complex. Importantly, the sites of APC cleavage at R306 and R506 leading to fVa inactivation are 60% exposed to solvent (E-F), unlike what is seen in fV (B-C). The structures also provide context for the functional consequences of posttranslational modifications and mutations in the C1 and C2 domains that reduce binding of fV to phospholipid membranes and perturb its cofactor activity. The site of glycosylation at N2181 and residue A2086 are clearly visible on the surface of the C2 domain, but residue W1920 is buried under Y1903 in the C1 domain (A,D).](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/137/22/10.1182_blood.2021010684/1/m_bloodbld2021010684f2.png?Expires=1769316358&Signature=DE-kyH9irKT288AITKYSkI8SCA07v2W67NS1kNhtj74terTh1UJuDcRjvvi3xpXW1xNJXSI70bN4AZuEl4gq8eIxbcHNfdjyKYRR1rVORju1fMJORobmadIjXuZqnFNxqdos0v34gGgd-QeZcnyxbMKtSdZ6vqvmbLWR7BuOWXOY0lfIENI7EHCwkmiCNSD7d2QTPYWGVxv6eCHYdDksgrFdQAGUfPrOm~Pcbhuz7rcukt8cRuNrFzcr~mEcVM8ODdz0DU87eQ052qgNqHtWPRCQzfgwFNZrxwKbssntzsNiX~O3C3UPUjJYH0LqdsqGB3oPCBsyBb1~Vy2z6yaHDA__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)