Key Points
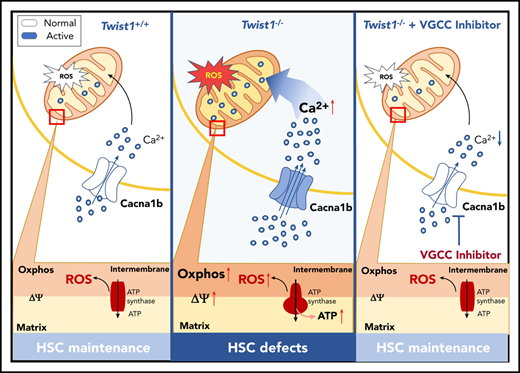
TWIST1 is required for HSC maintenance at steady state and protects HSCs against genotoxic stresses.
TWIST1 preserves the function of HSCs by repressing the CACNA1B/Ca2+/mitochondria axis.

Abstract
Mitochondria of hematopoietic stem cells (HSCs) play crucial roles in regulating cell fate and preserving HSC functionality and survival. However, the mechanism underlying HSC regulation remains poorly understood. Here, we identify transcription factor TWIST1 as a novel regulator of HSC maintenance through modulation of mitochondrial function. We demonstrate that Twist1 deletion results in significantly decreased lymphoid-biased HSC frequency, markedly reduced HSC dormancy and self-renewal capacity, and skewed myeloid differentiation in steady-state hematopoiesis. Twist1-deficient HSCs are more compromised in tolerance of irradiation- and 5-fluorouracil–induced stresses and exhibit typical phenotypes of senescence. Mechanistically, Twist1 deletion induces transactivation of voltage-gated calcium channel (VGCC) Cacna1b, which exhausts lymphoid-biased HSCs, impairs genotoxic hematopoietic recovery, and enhances mitochondrial calcium levels, metabolic activity, and reactive oxygen species production. Suppression of VGCC by a calcium channel blocker largely rescues the phenotypic and functional defects in Twist1-deleted HSCs under both steady-state and stress conditions. Collectively, our data, for the first time, characterize TWIST1 as a critical regulator of HSC function acting through the CACNA1B/Ca2+/mitochondria axis and highlight the importance of Ca2+ in HSC maintenance. These observations provide new insights into the mechanisms for the control of HSC fate.
Introduction
Hematopoietic stem cells (HSCs) maintain the hematopoietic system through their dual ability to self-renew and differentiate into progenitors of various lineages over the lifetime of an organism.1,2 At steady state, a majority of HSCs are maintained in a state of dormancy. After hematologic stress, HSCs are recruited from quiescence to rapidly repopulate a depleted hematopoietic compartment and restore homeostasis.3-6 Therefore, the ability to respond to stress is a fundamental determinant of HSCs for the prevention of stem cell exhaustion and bone marrow (BM) failure. Steady-state and stress hematopoieses are exquisitely regulated; however, their molecular processes remain incompletely characterized.
Mitochondria act as bioenergetic and biosynthetic hotspots that regulate various metabolic processes7 and play crucial roles in HSC homeostasis. At steady state, HSCs maintain low mitochondrial biogenesis and metabolic activity and rely on glycolysis during quiescence.8,9 During activation and differentiation, HSCs undergo a metabolic switch to oxidative phosphorylation (OXPHOS) and produce elevated levels of ATP and reactive oxygen species (ROS) to drive HSCs into the cell cycle, leading to reduced quiescence and impaired long-term reconstitution ability.10-12 The key role of mitochondria in the determination of HSC fate highlights the importance of understanding mitochondrial regulation. Recent studies have shown that low intracellular calcium is important for HSC maintenance in vitro,13 and the Ca2+/mitochondria pathway plays a crucial role in the regulation of HSC division.14 Given that intracellular Ca2+ is a critical regulator of mitochondrial energy metabolism,15 these data suggest the possible importance of Ca2+ in HSC fate decisions. However, to date, the role of Ca2+ in steady-state and stress hematopoieses and its modulation in HSCs remain largely unknown.
TWIST1, a highly conserved basic helix-loop-helix transcription factor, plays a pivotal role in mesoderm formation and myogenesis.16,17 Twist1 knockout (KO) mice die at embryonic day ∼11.5 with vascular and cranial neural tube defects.18 In adult tissue, Twist1 is preferentially expressed in mesodermally derived stem cells, such as muscle stem cells,19 mesenchymal stem cells (MSCs),20 and HSCs.21 The function of TWIST1 in the self-renewal and differentiation of muscle stem cell and MSCs has been well characterized22,23 ; however, its role in HSC maintenance under steady-state and stress conditions remains elusive.
To understand this, we generated a conditional KO (cKO) mouse model via Twist1 deletion in the adult hematologic system. Our data, for the first time, demonstrate that TWIST1 is required for HSC functionality through repression of the CACNA1B/Ca2+/mitochondria axis.
Materials and methods
Mice
Twist1flox/flox mice were purchased from the Mutant Mouse Regional Resource Center. Eight- to 12-week-old C57BL/6J-Ly5.1 or -Ly5.2 congenic mice were purchased from the animal facility of the State Key Laboratory of Experimental Hematology. Twist1flox/flox;Mx1-Cre mice were generated by crossing Twist1flox/flox mice with Mx1-Cre mice. Cre expression was induced by 4 consecutive intraperitoneal injections of polyinosinic:polycytidilic acid (5 mg/kg; GE Healthcare, Little Chalfont, United Kingdom) administered every other day. Prx1-CreER mice were obtained from The Jackson Laboratory. To induce Twist1flox/flox;Prx1-CreER mice, tamoxifen (Sigma-Aldrich, St Louis, MO) was injected intraperitoneally at a dose of 0.1 mg/g per injection for 5 consecutive days. All animal care and experimental procedures complied with the animal care guidelines approved by the Institutional Animal Care and Use Committees of State Key Laboratory of Experimental Hematology.
Evaluation of intracellular and mitochondrial Ca2+ levels, ROS levels, and mitochondrial properties
Intracellular Ca2+, mitochondrial Ca2+, mitochondrial membrane potential, and glucose uptake in HSCs were measured according to the manufacturers’ instructions using Fluo-4, AM (Thermo Fisher Scientific, Grand Island, NY), Rhod-2, AM, DilC1(5), and TMRM, and 2-NBDG (all from Thermo Fisher Scientific), respectively. For evaluation of mitochondrial mass, cells were incubated with MTG (Thermo Fisher Scientific) in the presence of verapamil24 (10 μM; Sigma-Aldrich) at 37°C for 30 min. ROS levels were evaluated by staining cells with DCFH-DA (Sigma-Aldrich) or CellROX Deep Red (Thermo Fisher Scientific). After staining, fluorescence intensity was determined via flow cytometry.
ATP measurement
The ATP concentration in sorted HSCs was measured using the ATP Determination Kit (Thermo Fisher Scientific).
Seahorse assays
The oxygen consumption rate was measured using XFe24 Extracellular Flux Assay Kits (Agilent Technologies, Santa Clara, CA) according to the manufacturer’s protocol. Briefly, 2 × 105 Lineage−Sca1+cKit+ (LSK) cells per well were seeded in a Cell-Tak–coated (Corning, New York, NY) 24-well XFe24 plate (Agilent Technologies). One hour before analysis, the medium was replaced with Seahorse XF media (Agilent Technologies). Analyses were performed at basal conditions and after injection of 1 µM of oligomycin (Agilent Technologies), 1 µM of FCCP (Agilent Technologies), and 0.5 µM of rotenone/antimycin A (Agilent Technologies).
Statistical analysis
All data are expressed as means ± standard deviations and are representative of at least 2 trials, with *P < .05, **P < .01, and ***P < .001. Statistical significance was calculated using a 2-tailed, unpaired or paired, Student t test. In survival experiments, mouse survival rates were analyzed using the log-rank test. Details of other experimental procedures are described in the supplemental Methods (available on the Blood Web site).
Data availability
RNA sequencing (RNA-seq) and ATAC-Seq data have been deposited in the National Center for Biotechnology Information Gene Expression Omnibus (accession GSE133682).
Results
Loss of Twist1 impairs HSC homeostasis
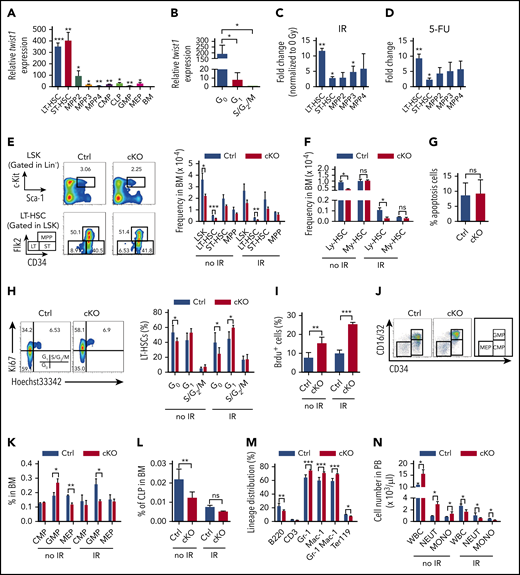
Our previous study has shown that Twist1 is highly enriched in HSCs.21 To further characterize the profiles of Twist1 expression in subdivided HSC populations and committed progenitor cells under steady-state and stress conditions, we performed quantitative reverse transcription polymerase chain reaction assays. The analysis showed that Twist1 expression was most abundant in long-term HSCs (LT-HSCs) and short-term HSCs (ST-HSCs; Figure 1A). LT-HSCs in the G0 phase exhibited the highest Twist1 levels compared with those in the G1 and S/G2/M phases (Figure 1B). The most significant upregulation of Twist1 was observed in HSCs when HSCs and MPPs were treated with IR and 5-FU (Figure 1C-D). These expression patterns imply an important role of TWIST1 in regulating HSC function.
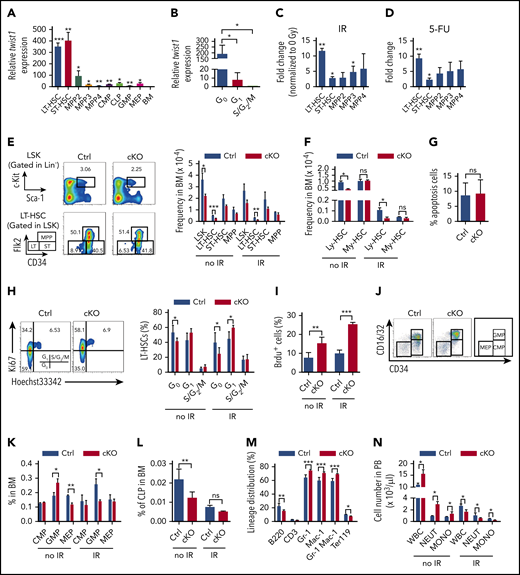
HSC homeostasis is impaired in Twist1-deficient mice. (A) Relative expression levels of Twist1 in different hematopoietic cell subsets were evaluated by quantitative polymerase chain reaction. Each subset was compared with BM for statistical analysis (n = 3). LT-HSC: Flk2−CD150+CD48−LSK; ST-HSC: Flk2−CD150−CD48−LSK; multipotent progenitor 2 (MPP2): Flk2−CD150+CD48+LSK; MPP3: Flk2−CD150−CD48+LSK; MPP4: Flk2+CD150−CD48+LSK; common myeloid (My) progenitor (CMP): CD34+CD16/32−LK (Lineage−cKit+Sca1−); granulocyte/monocyte progenitor (GMP): CD34+CD16/32+LK; megakaryocyte/erythrocyte progenitor (MEP): CD34−CD16/32−LK; CLP: CD127+cKitlowSca1low Lineage−. (B) Relative expression levels of Twist1 messenger RNA (mRNA) in CD34−Flk2− LT-HSCs in G0, G1, and S/G2/M phases (n = 3). (C) LT-HSCs, ST-HSCs, and MPP2-4 cells were isolated from mice before and at 12 hours after irradiation (IR; 5 Gy). Twist1 mRNA was detected, and the fold change of Twist1 expression was normalized to the 0-Gy group (n = 3). (D) Induced expression of Twist1 mRNA in LT-HSCs, ST-HSCs, and MPP2-4 cells 10 days after 5-fluorouracil (5-FU) treatment. Fold change of Twist1 expression was normalized to the no-treatment group (n = 3). (E) Frequencies of indicated populations in BM cells from control (Ctrl) and cKO mice at 4 weeks after the last polyinosinic:polycytidilic acid injection and day 12 after IR (right) and representative fluorescence-activated cell sorting (FACS) plots (left; n = 5). (F) Percentages of CD150highLSKFlk2−CD48− and CD150lowLSKFlk2−CD48− cells in BM cells from Ctrl and cKO mice at steady state and day 12 after IR (n = 3-5). (G) Apoptosis in BM CD150low HSCs under steady-state conditions (n = 4). (H) Cell-cycle analysis of LT-HSCs at basal and day 12 post-IR (right) and representative FACS plots (left; n = 5). (I) Proliferation analysis of LT-HSCs at basal and day 12 post-IR (n = 4). (J-K) Frequencies of CMPs, GMPs, and MEPs in BM cells at steady state and day 12 after IR (K) and representative FACS plots (J) (n = 5). (L) Frequencies of common lymphoid (Ly) progenitors (CLPs) in BM under steady-state conditions and after IR (n = 5). (M) Lineage distribution in BM assessed by FACS at steady state (n = 5). (N) Absolute number per liter of white blood cells (WBCs), neutrophils, and monocytes in peripheral blood (PB) from Ctrl and cKO mice under steady-state conditions and after IR (n = 4-5). Data are represented as means ± standard deviation. *P < .05, **P < .01, ***P < .001 (Student t test). ns, not significant.
HSC homeostasis is impaired in Twist1-deficient mice. (A) Relative expression levels of Twist1 in different hematopoietic cell subsets were evaluated by quantitative polymerase chain reaction. Each subset was compared with BM for statistical analysis (n = 3). LT-HSC: Flk2−CD150+CD48−LSK; ST-HSC: Flk2−CD150−CD48−LSK; multipotent progenitor 2 (MPP2): Flk2−CD150+CD48+LSK; MPP3: Flk2−CD150−CD48+LSK; MPP4: Flk2+CD150−CD48+LSK; common myeloid (My) progenitor (CMP): CD34+CD16/32−LK (Lineage−cKit+Sca1−); granulocyte/monocyte progenitor (GMP): CD34+CD16/32+LK; megakaryocyte/erythrocyte progenitor (MEP): CD34−CD16/32−LK; CLP: CD127+cKitlowSca1low Lineage−. (B) Relative expression levels of Twist1 messenger RNA (mRNA) in CD34−Flk2− LT-HSCs in G0, G1, and S/G2/M phases (n = 3). (C) LT-HSCs, ST-HSCs, and MPP2-4 cells were isolated from mice before and at 12 hours after irradiation (IR; 5 Gy). Twist1 mRNA was detected, and the fold change of Twist1 expression was normalized to the 0-Gy group (n = 3). (D) Induced expression of Twist1 mRNA in LT-HSCs, ST-HSCs, and MPP2-4 cells 10 days after 5-fluorouracil (5-FU) treatment. Fold change of Twist1 expression was normalized to the no-treatment group (n = 3). (E) Frequencies of indicated populations in BM cells from control (Ctrl) and cKO mice at 4 weeks after the last polyinosinic:polycytidilic acid injection and day 12 after IR (right) and representative fluorescence-activated cell sorting (FACS) plots (left; n = 5). (F) Percentages of CD150highLSKFlk2−CD48− and CD150lowLSKFlk2−CD48− cells in BM cells from Ctrl and cKO mice at steady state and day 12 after IR (n = 3-5). (G) Apoptosis in BM CD150low HSCs under steady-state conditions (n = 4). (H) Cell-cycle analysis of LT-HSCs at basal and day 12 post-IR (right) and representative FACS plots (left; n = 5). (I) Proliferation analysis of LT-HSCs at basal and day 12 post-IR (n = 4). (J-K) Frequencies of CMPs, GMPs, and MEPs in BM cells at steady state and day 12 after IR (K) and representative FACS plots (J) (n = 5). (L) Frequencies of common lymphoid (Ly) progenitors (CLPs) in BM under steady-state conditions and after IR (n = 5). (M) Lineage distribution in BM assessed by FACS at steady state (n = 5). (N) Absolute number per liter of white blood cells (WBCs), neutrophils, and monocytes in peripheral blood (PB) from Ctrl and cKO mice under steady-state conditions and after IR (n = 4-5). Data are represented as means ± standard deviation. *P < .05, **P < .01, ***P < .001 (Student t test). ns, not significant.
To verify this, we bred Twist1flox/flox mice with the Mx1-Cre strain to ablate Twist1 in the adult hematopoietic system. Twist1 was efficiently deleted from the hematopoietic system after polyinosinic:polycytidilic acid administration (supplemental Figure 1A-B). The expression levels of Twist2 in Twist1-deleted hematopoietic stem/progenitor cells (HSPCs) did not increase to compensate (supplemental Figure 1C). We observed that BM cellularity and spleen weight were not altered in Twist1 cKO mice as compared with control mice (supplemental Figure 1D-E). The frequencies and numbers of CD34−Flk2− LT-HSCs and LSK cells were significantly reduced (by 2.0- and 1.6-fold in frequency, respectively) in Twist1-deficient BM (Figure 1E; supplemental Figure 1F). Interestingly, the myeloid- and lymphoid-biased HSCs, distinguished by CD150,25 were not equivalently reduced. The frequency of lymphoid-biased HSCs in Twist1-deficient mice was prominently decreased (by 3.0-fold), whereas the reduction in myeloid-biased HSCs was not statistically significant (by 1.2-fold; Figure 1F). The reduction in the lymphoid-biased HSC compartment of Twist1 cKO mice was not due to enhanced cell death (Figure 1G). Additionally, Twist1 cKO mice showed markedly decreased frequencies of LT-HSCs, ST-HSCs, and MPPs in the spleen (supplemental Figure 1G) and noticeably reduced frequency of LSKs in the PB (supplemental Figure 1H).
We next analyzed the cell-cycle status of BM LT-HSCs using Ki67 staining. Twist1 cKO mice exhibited a noticeable decrease in the percentage of LT-HSCs in the G0 phase (41% in cKO vs 53% in control mice; Figure 1H). Additionally, the fraction of nucleotide analog bromodeoxyuridine–incorporated LT-HSCs was significantly increased in Twist1-deleted mice (Figure 1I), suggesting enhanced cell division. The defect in quiescence of Twist1-deficient LT-HSCs was further supported by our observation that the expression of Cdkn1c, Cdkn1a, and Cdkn1b cyclin-dependent kinase inhibitors regulating HSC dormancy26 was considerably downregulated in these cells (supplemental Figure 1I).
Evaluation of BM progenitor populations indicated a significant increase in the frequency and number of granulocyte/monocyte progenitors, whereas the proportion and number of megakaryocyte/erythrocyte progenitors, as well as common lymphoid progenitors, were markedly reduced (Figure 1J-L; supplemental Figure 1J-K). Within the more mature compartment in the BM, Twist1 deletion resulted in a prominent elevation in myeloid cells and significant reductions in lymphoid and erythroid cells (Figure 1M). In the spleen, Twist1 deletion led to slight decreases in common lymphoid progenitors and T lymphoid cells, whereas the frequencies of other progenitor and mature fractions were unaffected (supplemental Figure 1L-N). Analysis of PB revealed a marked increase in white blood cell counts, particularly neutrophils and monocytes (Figure 1N). These data suggest that Twist1 deletion causes enhanced myeloid lineage differentiation in both the BM and periphery, and the myeloid skewing may be due to the reduction in lymphoid-biased HSCs.
Collectively, these observations support an essential role for TWIST1 in maintaining lymphoid-biased HSC pool size and quiescence and fidelity of differentiation in steady-state hematopoiesis.
Twist1 deletion leads to reduced reconstitution and self-renewal capacity of HSCs
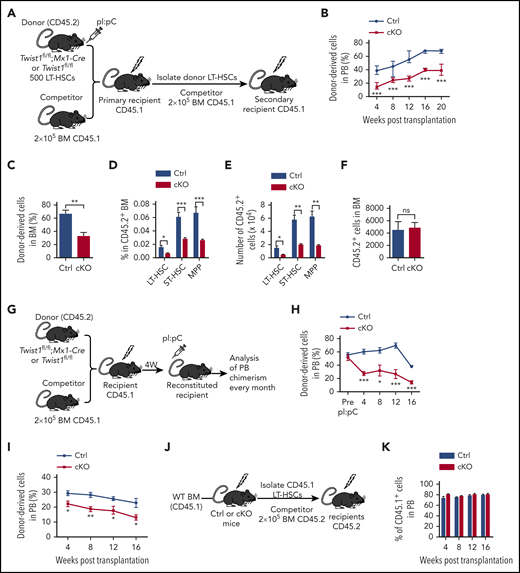
To assess whether the HSCs in Twist1-deleted mice had a functional defect, we performed a competitive reconstitution assay (Figure 2A). Donor-derived PB chimerism of Twist1-deleted cell transplant recipients was significantly lower than that of controls at 4 weeks posttransplantation (Figure 2B). Twenty weeks after transplantation, BM chimerism exhibited the same trend as PB (Figure 2C), and these mice showed significant decreases in the frequency and total number of donor-derived LT-HSCs, ST-HSCs, and MPPs (Figure 2D-E). Importantly, the reconstitution defect of Twist1-deficient HSCs was not due to a defect in BM homing (Figure 2F). When Twist1 deletion was induced after HSC engraftment in recipient mice (Figure 2G), similar to that induced before transplantation, a significantly competitive defect was also observed (Figure 2H). These results suggest that Twist1 deletion reduces the capacity of the in vivo competitive repopulation of HSCs. Moreover, the differentiation bias toward the myeloid lineage was reproducible at 20 weeks after transplantation (supplemental Figure 2A-D), confirming that Twist1-deficient HSCs harbored enhanced myeloid potential.
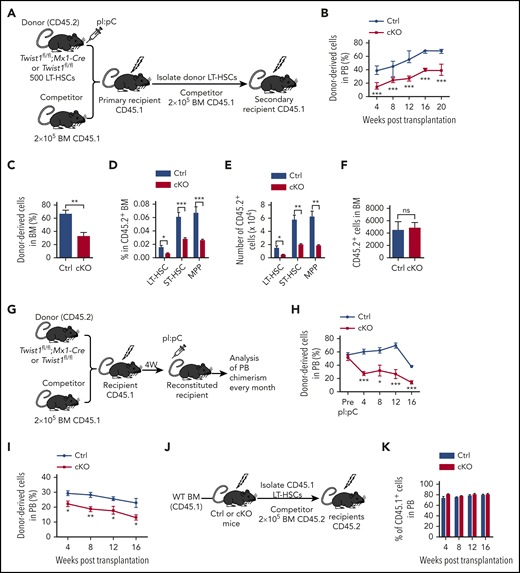
Twist1 deletion compromises reconstitution and self-renewal capacity of HSCs. (A) Schematic for competitive and secondary BM transplantations. (B) Percentage of donor-derived CD45.2 chimerism in the PB of primary recipients. Excision occurred 3 weeks before transplantation (n = 5-10). (C) Chimerism of donor-derived BM cells at 20 weeks after primary BM transplantation (BMT; n = 5). (D-E) Frequencies and absolute numbers of donor-derived indicated populations in CD45.2+ BM at 20 weeks after primary BMT (n = 5). (F) Homing assay was performed by transplantation of 5000 LT-HSCs from control (Ctrl) or cKO mice into lethally irradiated wild-type (WT) mice. Absolutes number of CD45.2+ cells homing to recipient BM are shown (n = 5). (G) Schematic for competitive BMTs. (H) Percentage of CD45.2 chimerism in the PB of primary recipients after gene deletion in recipient mice (n = 4-6). (I) Chimerism of donor-derived PB cells after secondary BMT (n = 4-6). CD45.2+ LT-HSCs from primary BMT mice were transplanted into lethally irradiated secondary recipients. (J) Schematic for reciprocal BMTs. (K) Chimerism of CD45.1+ cells in PB of cKO and Ctrl recipients at the indicated time points after BMT (n = 8-9). Data are shown as means ± standard deviation. *P < .05, **P < .01, ***P < .001 (Student t test). ns, not significant; pI:pC, polyinosinic:polycytidilic acid.
Twist1 deletion compromises reconstitution and self-renewal capacity of HSCs. (A) Schematic for competitive and secondary BM transplantations. (B) Percentage of donor-derived CD45.2 chimerism in the PB of primary recipients. Excision occurred 3 weeks before transplantation (n = 5-10). (C) Chimerism of donor-derived BM cells at 20 weeks after primary BM transplantation (BMT; n = 5). (D-E) Frequencies and absolute numbers of donor-derived indicated populations in CD45.2+ BM at 20 weeks after primary BMT (n = 5). (F) Homing assay was performed by transplantation of 5000 LT-HSCs from control (Ctrl) or cKO mice into lethally irradiated wild-type (WT) mice. Absolutes number of CD45.2+ cells homing to recipient BM are shown (n = 5). (G) Schematic for competitive BMTs. (H) Percentage of CD45.2 chimerism in the PB of primary recipients after gene deletion in recipient mice (n = 4-6). (I) Chimerism of donor-derived PB cells after secondary BMT (n = 4-6). CD45.2+ LT-HSCs from primary BMT mice were transplanted into lethally irradiated secondary recipients. (J) Schematic for reciprocal BMTs. (K) Chimerism of CD45.1+ cells in PB of cKO and Ctrl recipients at the indicated time points after BMT (n = 8-9). Data are shown as means ± standard deviation. *P < .05, **P < .01, ***P < .001 (Student t test). ns, not significant; pI:pC, polyinosinic:polycytidilic acid.
To assess the impact of Twist1 deletion on HSC self-renewal potential, we performed secondary transplantation 20 weeks after the first transplantation. We observed much lower PB chimerism in the Twist1-deleted cell transplantation group, suggesting that TWIST1 is critical for HSC self-renewal (Figure 2I).
Twist1 has been reported to be highly expressed in MSCs.22 To exclude nonhematopoietic contributions to the observed phenotype, we performed reciprocal BM transplantation. WT LT-HSCs were transplanted into cKO or control mice (Figure 2J). The results showed comparable HSC reconstitution and multilineage differentiation between the 2 groups (Figure 2K; supplemental Figure 3A). No alterations were found in PB or BM chimerism after secondary transplantation (supplemental Figure 3B-C). To further exclude the possible cell-extrinsic effects of TWIST1 from MSCs, we used Prx1-CreER;Twist1flox/flox mice to explore whether deletion of Twist1 in mesenchymal stem/progenitor cells has an impact on HSCs. The results showed that the frequencies of HSPCs in the BM and spleen were comparable between control and Twist1-deficient mice (supplemental Figure 4A-E). In addition, the differentiation status in the BM, PB, and spleen of Twist1 cKO mice was also unaffected (supplemental Figure 4F-H). These observations indicate that Twist1 depletion in MSCs does not affect HSCs and that Twist1 KO mediated by Mx1-Cre impairs the phenotype and functions of HSCs in a cell-intrinsic manner.
Twist1 deficiency results in impaired hematopoiesis upon genotoxic stresses
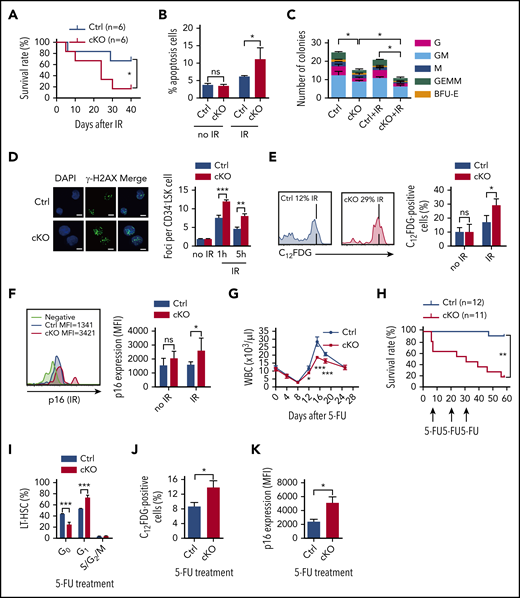
We further investigated how Twist1-deficient HSCs respond to genotoxic stresses, including IR and the chemotherapeutic agent 5-FU. First, Twist1 cKO and control animals were exposed to γ-IR at a high dose of 7 Gy; 67% of the control mice survived beyond 40 days, whereas the cKO mice died at a median of 18 days (Figure 3A). To substantiate these data, we transplanted Twist1-deleted or control BM cells into WT recipient mice. After BM reconstitution, the animals underwent IR at 5 Gy. Mice reconstituted with Twist1-deleted cells died much faster than controls (supplemental Figure 5A). These findings suggest that TWIST1 protects mice from IR-induced death.
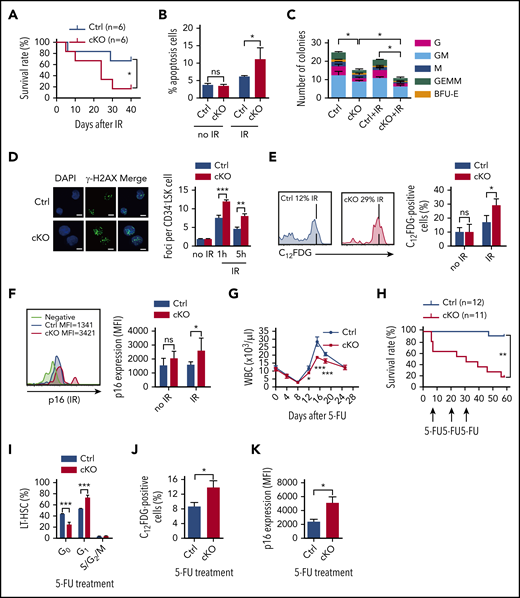
Twist1 cKO animals and HSCs are sensitized to IR and 5-FU. (A) Kaplan-Meier survival curves of mice after IR at 7 Gy (n = 6; log-rank test). (B) Frequencies of apoptotic cells in the LT-HSCs from mice at basal and 12 days after IR (n = 4; Student t test). (C) Colony-forming capacity of LT-HSCs. LT-HSCs from cKO and control (Ctrl) mice, untreated (no IR) or after IR (2 Gy), were plated in methylcellulose-containing media, and colonies were counted and classified as GEMM, GM, G, M, or BFU-E on day 10 of culture (n = 4; Student t test). (D) Quantification of γ-H2AX foci (green) in nuclei (blue) of CD34− LSK cells without IR and at the indicated times post-IR at 2 Gy (right) and representative confocal microscopic images at 1 hr after IR (left). Scale bars, 10 µm (Student t test). (E) Percentages of SA-β-gal staining in LT-HSCs at basal and 12 days after IR at 5 Gy (right) and representative fluorescence-activated cell sorting (FACS) plots (left; n = 4-5; Student t test). (F) The MFI of p16 expression in LT-HSCs under steady-state conditions and after IR and representative FACS plots (left; n = 5-6; Student t test). (G) Absolute leukocyte count in PB after a single injection of 5-FU (n = 4-6; Student t test). (H) Kaplan-Meier survival curves of mice after serial doses of 5-FU treatment at 10-day intervals (n = 11-12; log-rank test). (I) Cell-cycle analysis of LT-HSCs in BM after 5-FU treatment (n = 4; Student t test). (J) Percentages of SA-β-gal staining in LT-HSCs at day 16 after 5-FU injection (n = 5). (K) The MFI of p16 expression in LT-HSCs after 5-FU treatment (n = 5). Data are shown as means ± standard deviation. *P < .05, **P < .01, ***P < .001. DAPI, 4′,6-diamidino-2-phenylindole; MFI, mean fluorescence intensity; ns, not significant; WBC, white blood cell.
Twist1 cKO animals and HSCs are sensitized to IR and 5-FU. (A) Kaplan-Meier survival curves of mice after IR at 7 Gy (n = 6; log-rank test). (B) Frequencies of apoptotic cells in the LT-HSCs from mice at basal and 12 days after IR (n = 4; Student t test). (C) Colony-forming capacity of LT-HSCs. LT-HSCs from cKO and control (Ctrl) mice, untreated (no IR) or after IR (2 Gy), were plated in methylcellulose-containing media, and colonies were counted and classified as GEMM, GM, G, M, or BFU-E on day 10 of culture (n = 4; Student t test). (D) Quantification of γ-H2AX foci (green) in nuclei (blue) of CD34− LSK cells without IR and at the indicated times post-IR at 2 Gy (right) and representative confocal microscopic images at 1 hr after IR (left). Scale bars, 10 µm (Student t test). (E) Percentages of SA-β-gal staining in LT-HSCs at basal and 12 days after IR at 5 Gy (right) and representative fluorescence-activated cell sorting (FACS) plots (left; n = 4-5; Student t test). (F) The MFI of p16 expression in LT-HSCs under steady-state conditions and after IR and representative FACS plots (left; n = 5-6; Student t test). (G) Absolute leukocyte count in PB after a single injection of 5-FU (n = 4-6; Student t test). (H) Kaplan-Meier survival curves of mice after serial doses of 5-FU treatment at 10-day intervals (n = 11-12; log-rank test). (I) Cell-cycle analysis of LT-HSCs in BM after 5-FU treatment (n = 4; Student t test). (J) Percentages of SA-β-gal staining in LT-HSCs at day 16 after 5-FU injection (n = 5). (K) The MFI of p16 expression in LT-HSCs after 5-FU treatment (n = 5). Data are shown as means ± standard deviation. *P < .05, **P < .01, ***P < .001. DAPI, 4′,6-diamidino-2-phenylindole; MFI, mean fluorescence intensity; ns, not significant; WBC, white blood cell.
To assess the HSC recovery potential, we exposed cKO and control mice to IR at 5 Gy. The cKO mice displayed significant decreases in total white blood cell counts and BM cellularity at day 12 post-IR compared with controls (Figure 1N; supplemental Figure 5B). Evaluation of BM HSPCs revealed that the frequency of LT-HSCs in Twist1-deficient mice was prominently decreased upon IR stress (by 2.8-fold; Figure 1E). The rate of apoptosis in LT-HSCs was significantly increased (Figure 3B), consistent with the markedly decreased expression of antiapoptotic Bcl2 and MnSOD (supplemental Figure 5C), suggesting that the reduced HSC number was related to cell viability. Moreover, Twist1-deficient LT-HSCs were less quiescent (24% in cKO vs 40% in control mice; Figure 1H) and had a higher proportion of proliferating cells (Figure 1I) upon IR stress. The extent of reduction in the number and dormancy of LT-HSCs caused by Twist1 KO under IR was more pronounced than that under steady-state conditions. Moreover, the fraction of granulocyte/monocyte progenitors was significantly reduced in Twist1 cKO mice, despite its prominent increase under steady-state conditions (Figure 1K). Functionally, Twist1-deleted LT-HSCs showed a dramatic decrease in colony-forming ability in methylcellulose (Figure 3C), and the decrease was more significant after exposure to IR at 2 Gy (Figure 3C).
We next tested whether the increased sensitivity of the HSCs to IR was accompanied by DNA damage accumulation. We observed that DNA damage–induced γ-H2AX foci were significantly aggravated in Twist1-deleted HSCs after IR (Figure 3D). This result was further confirmed via an alkaline comet assay (supplemental Figure 5D). Accumulation of DNA damage is often associated with cellular senescence.27 As anticipated, Twist1-deleted LT-HSCs upon IR treatment displayed significantly elevated SA-β-galactosidase activity and p16INK4a expression, both of which are markers of senescence (Figure 3E-F), whereas a typical senescent phenotype was not observed under steady-state conditions (Figure 3E-F). It has been reported that p16INK4a causes hypophosphorylation of Rb, which is essential for inducing the senescent phenotype.28,29 We demonstrated that the expression of pRb was increased and hypophosphorylated pRb was reduced in Twist1-deleted LT-HSCs after IR treatment, further confirming the existence of senescence (supplemental Figure 5E). Collectively, these findings suggest that TWIST1 is essential for protecting HSCs from IR-induced apoptosis, senescence, and DNA damage accumulation.
Intriguingly, overall similar effects could be observed when Twist1 cKO and control mice were challenged with the myeloablative agent 5-FU, which preferentially eliminates actively cycling cells. First, Twist1 cKO mice exhibited markedly delayed hematopoietic recovery, as reflected in multilineage defects in the generation of mature cells in PB, after a single 5-FU injection (Figure 3G; supplemental Figure 6A). Moreover, the BM cellularity was significantly reduced (supplemental Figure 6B) and the recovery of LT-HSCs, ST-HSCs, and MPPs was impaired in Twist1-deficient mice (supplemental Figure 6C). These results prompted us to investigate the sensitivity of Twist1 cKO mice to persistent 5-FU treatment, which induces HSC exhaustion.30 Although 92% of control mice survived, only 25% of the Twist1-deficient mice did so (Figure 3H), implying that Twist1-deficient HSCs were less quiescent. Consistent with this, the percentage of Twist1-deleted LT-HSCs in the G0 phase was significantly decreased upon 5-FU treatment (23% in cKO vs 43% in control mice; Figure 3I), and the extent of reduction was more pronounced than that under steady-state conditions. Finally, although comparable levels of γ-H2AX foci were detected (data not shown), the senescent phenotype was markedly increased in Twist1-deleted HSCs in comparison with controls (Figure 3J-K; supplemental Figure 6D).
Notably, when HSCs were subdivided into lymphoid- and myeloid-biased HSCs, the frequencies of lymphoid-biased HSCs in Twist1 cKO mice were observed to be remarkably decreased upon both IR and 5-FU treatments, and moreover, the reductions in lymphoid-biased HSCs were more prominent than in LT-HSCs (Figure 1E-F and supplemental Figure 6C,E, respectively). In contrast, the proportions of Twist1-defecient myeloid-biased HSCs remained unchanged under these stress conditions (Figure 1F and supplemental Figure 6E, respectively). Taken together, these results indicate that TWIST1 protects lymphoid-biased, but not myeloid-biased, HSCs from exhaustion and facilitates hematopoietic recovery against genotoxic stresses.
Twist1 deficiency leads to altered expression of genes associated with regulation of Ca2+ and mitochondrial function in LT-HSCs upon IR treatment
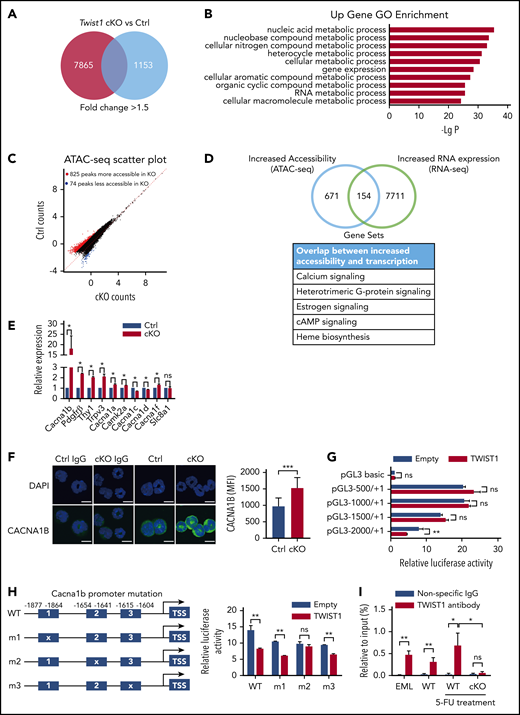
To explore the molecular mechanisms underlying the effect of TWIST1 on HSCs upon stress, we performed RNA-seq on Twist1-deleted LT-HSCs and controls after IR treatment. A total of 9018 genes were found to be differentially expressed (7865 upregulated and 1153 downregulated; >1.5-fold change; Figure 4A). Gene ontology analysis of the upregulated cohort revealed that metabolic genes were most significantly enriched (Figure 4B). Gene set enrichment analysis showed that voltage-gated calcium channel (VGCC) and calcium ion transmembrane transporter activity were activated in Twist1-deficient LT-HSCs (Table 1). The negative modulators of mitochondrial function, such as Hif1a, Foxo3, Atm, Atr, and Tsc1,31-34 were enriched significantly within downregulated genes in Twist1-deficient LT-HSCs (supplemental Table 1). These data suggest that calcium signaling and mitochondrial function might be hyperactivated in these cells upon IR stress.
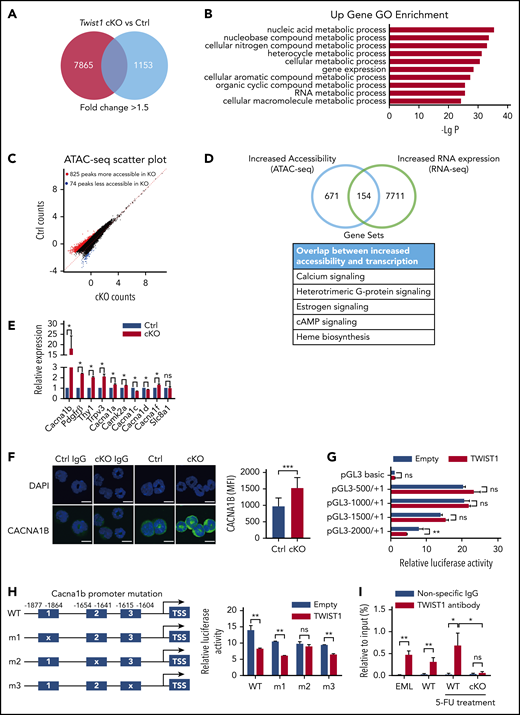
Twist1 deletion displays altered expression of genes associated with regulation of Ca2+ and mitochondrial function upon IR stress. (A) Transcriptome profiling of cKO and control (Ctrl) LT-HSCs after IR treatment. Red box displays the number of upregulated genes, and blue box displays the number of downregulated genes. (B) Gene ontology (GO) analysis of genes upregulated >1.5-fold in Twist1-deficient LT-HSCs after IR stress. Only top 10 GO terms are listed. (C) Scatterplot shows differentially accessible peaks from ATAC-seq analysis between cKO and Ctrl LT-HSCs after IR stress. (D) Venn diagram of genes with discrete Twist1 peaks compared with genes upregulated after Twist1 deletion. (E) Relative messenger RNA expression levels of calcium signaling factors in LT-HSCs from cKO and Ctrl BM (n = 3; Student t test). (F) Quantification of CACNA1B expression (green) in HSCs at 12 days after IR at 5 Gy (right) and representative confocal microscopic images (left). Rabbit immunoglobulin G (IgG) was used as a negative control. Scale bars, 10 µm (Student t test). (G) Promoter activity assay using a Twist1 expression plasmid and luciferase reporter constructs driven by various lengths of the Cacna1b 5′ flanking region in the 293T cell line (n = 3; Student t test). (H) Relative luciferase activity in 293T cells transfected with WT or mutated Cacna1b promoter–luciferase reporter constructs (right). The schematic representation of the mutated constructs of the Cacna1b promoter (left; n = 3; Student t test). (I) Binding of TWIST1 was analyzed by chromatin immunoprecipitation–polymerase chain reaction in EML cell line and LSK cells from WT mice or from Ctrl and cKO mice treated with 5-FU (n v= 3; Student t test). Column plots show means ± standard deviation. *P < .05, **P < .01, ***P < .001. ns, not significant.
Twist1 deletion displays altered expression of genes associated with regulation of Ca2+ and mitochondrial function upon IR stress. (A) Transcriptome profiling of cKO and control (Ctrl) LT-HSCs after IR treatment. Red box displays the number of upregulated genes, and blue box displays the number of downregulated genes. (B) Gene ontology (GO) analysis of genes upregulated >1.5-fold in Twist1-deficient LT-HSCs after IR stress. Only top 10 GO terms are listed. (C) Scatterplot shows differentially accessible peaks from ATAC-seq analysis between cKO and Ctrl LT-HSCs after IR stress. (D) Venn diagram of genes with discrete Twist1 peaks compared with genes upregulated after Twist1 deletion. (E) Relative messenger RNA expression levels of calcium signaling factors in LT-HSCs from cKO and Ctrl BM (n = 3; Student t test). (F) Quantification of CACNA1B expression (green) in HSCs at 12 days after IR at 5 Gy (right) and representative confocal microscopic images (left). Rabbit immunoglobulin G (IgG) was used as a negative control. Scale bars, 10 µm (Student t test). (G) Promoter activity assay using a Twist1 expression plasmid and luciferase reporter constructs driven by various lengths of the Cacna1b 5′ flanking region in the 293T cell line (n = 3; Student t test). (H) Relative luciferase activity in 293T cells transfected with WT or mutated Cacna1b promoter–luciferase reporter constructs (right). The schematic representation of the mutated constructs of the Cacna1b promoter (left; n = 3; Student t test). (I) Binding of TWIST1 was analyzed by chromatin immunoprecipitation–polymerase chain reaction in EML cell line and LSK cells from WT mice or from Ctrl and cKO mice treated with 5-FU (n v= 3; Student t test). Column plots show means ± standard deviation. *P < .05, **P < .01, ***P < .001. ns, not significant.
To further explore the regulatory elements involved in the IR-induced stress response of Twist1-deleted HSCs, we performed ATAC-seq. The results showed significant changes in chromatin accessibility, with a gain of 825 peaks and a loss of 74 accessible peaks in the Twist1-deficient LT-HSCs after IR treatment (Figure 4C). Notably, an integrative analysis of ATAC-seq and RNA-seq data revealed a significant correlation between increased chromatin accessibility and upregulated genes (supplemental Figure 7) and identified 154 overlapping genes (Figure 4D). Enrichment analysis of the overlapping genes revealed that calcium, heterotrimeric G-protein, cAMP, and heme biosynthesis signaling were significantly altered, all of which are associated with mitochondrial biogenesis and function. Of these, the calcium signaling pathway was increased most markedly (Figure 4D). Quantitative polymerase chain reaction and confocal fluorescence microscopy validation of genes involved in the calcium signaling pathway revealed that the expression levels of the members of the VGCC family calcium channel a1b subunit (Cacna1b) were most dramatically elevated (by 20-fold in messenger RNA) in Twist1-deleted HSCs after IR treatment (Figure 4E-F). Intriguingly, data mining of an available data set revealed that the expression of Cacna1b was downregulated in murine LT-HSCs and increased with differentiation (supplemental Figure 8A). We further demonstrated that LT-HSCs in the G0 phase showed the lowest Cacna1b levels compared with those in the G1 and S/G2/M phases (supplemental Figure 8B). These analyses suggest an inverse relationship between Cacna1b and Twist1 expression in HSCs. To determine whether TWIST1 directly regulates the transcription of Cacna1b, we first searched the JASPAR database and found an E-box consensus sequence, a well-defined TWIST1-binding motif, in the promoter region of Cacna1b. We then examined the promoter activity of various lengths of Cacna1b 5′ flanking sequences (0.5, 1, 1.5, and 2 kb) using a luciferase reporter assay. Only the 2-kb fragment showed a significant decrease in luciferase activity in response to Twist1 overexpression (Figure 4G), indicating that the segment spanning the −2- to −1.5-kb region is important for promoter activity. When the DNA sequence between −1654 and −1641 was mutated, the promoter activity was abrogated dramatically (Figure 4H). Binding of endogenous TWIST1 to the Cacna1b promoter was determined using chromatin immunoprecipitation assays in multipotent hematopoietic EML cell line and LSK cells isolated from WT mice under steady-state and 5-FU stress conditions (Figure 4I). In contrast, TWIST1 binding was impaired in LSK cells of cKO mice upon 5-FU treatment (Figure 4I). These results indicate that TWIST1 is a transcriptional repressor of CACNA1B under steady-state and stress conditions. Collectively, treatment of Twist1-deleted LT-HSCs with IR results in altered expression of genes related to the regulation of Ca2+ and mitochondrial function.
Twist1 deficiency impairs HSC function by activating CACNA1B and subsequent enhanced mitochondrial function
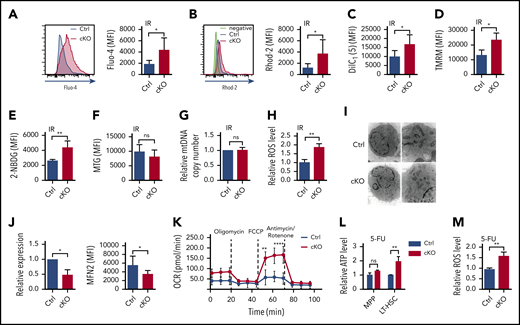
It has been reported that CACNA1B is a regulator of the intracellular Ca2+ level by influx of extracellular Ca2+ and is important for metabolic status.35 As expected, the upregulation of Cacna1b led to elevated intracellular Ca2+ concentration in Twist1-deficient LT-HSCs under IR stress by using Fluo-4 (Figure 5A). Because the upregulation of intracellular Ca2+ concentration triggers the mitochondrial Ca2+ level,36 which plays a critical role in boosting mitochondrial function, we next measured the mitochondrial properties of Twist1-deleted HSCs upon IR treatment. We found that the mitochondrial Ca2+ level determined via Rhod-2 staining and mitochondrial membrane potential (ΔΨm) evaluated by using DilC1(5) and TMRM probes were markedly elevated (Figure 5B-D). Moreover, a prominent increase in glucose uptake was observed as well in Twist1-deficient LT-HSCs using 2-NBDG staining (Figure 5E). No obvious difference in mitochondrial mass was observed between control and Twist1-deficient LT-HSCs by using MTG in the presence of verapamil (Figure 5F), and this result was confirmed by mitochondrial DNA quantification (Figure 5G). In addition, as a byproduct of OXPHOS, the level of ROS was evidently increased (Figure 5H). To further define whether Twist1 deficiency causes functional heterogeneity within the HSC pool, we examined calcium levels and mitochondrial properties of CD150high and CD150low HSCs upon IR treatment. Twist1 deletion led to increased intracellular and mitochondrial Ca2+ concentrations, mitochondrial membrane potential, and glucose uptake in lymphoid-biased HSCs, but not in myeloid-biased HSCs (supplemental Figure 9A-D). In summary, these results suggest TWIST1 restrains the activation of Ca2+ influx and mitochondrial function in lymphoid-biased HSCs after IR treatment.
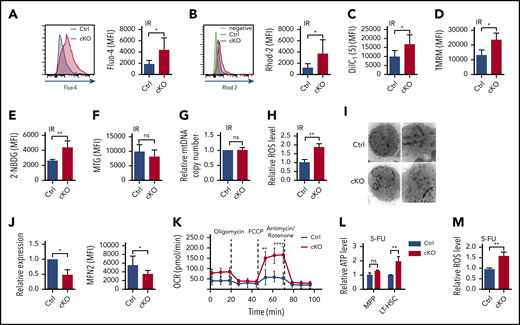
Loss of Twist1 enhances Ca2+ level, mitochondrial activity and metabolism, and ROS level in HSCs upon genotoxic stresses. (A-B) The concentrations of intracellular Ca2+ (A) and mitochondrial Ca2+ (B) in LT-HSCs after IR stress. Representative fluorescence-activated cell sorting (FACS) plots show the fluorescence intensity of Fluo-4 (intracellular Ca2+) and Rhod-2 (mitochondrial Ca2+; n = 5). (C) Mitochondrial membrane potential of LT-HSCs determined by DilC1(5) staining after IR (n = 5). (D) Mitochondrial membrane potential of LT-HSCs determined by TMRM staining after IR (n = 5). (E) Glucose uptake of LT-HSCs after IR (n = 5). (F) Mitochondrial mass of LT-HSCs evaluated by using MTG staining in the presence of verapamil after IR (n = 5). (G) Relative mitochondrial DNA (mtDNA) copy number of LSK cells evaluated by DNA quantitative polymerase chain reaction (PCR; n = 2). (H) Relative ROS levels of LT-HSCs after IR (n = 5). (I) Representative TEM images of mitochondrial morphology in LSK cells after 5-FU treatment (left, 10 000×; right, 20 000×). (J) Expression of Mfn2 messenger RNA based on quantitative reverse transcription PCR and MFN2 protein determined by FACS in LT-HSCs after 5-FU treatment (n = 3-5). (K) Oxygen-consumption rate (OCR) of LSK cells after 5-FU treatment (n = 4). (L) Relative ATP levels of MPPs and LT-HSCs after 5-FU treatment (n = 4). (M) Relative ROS levels of LT-HSCs after 5-FU treatment (n = 5). Data are shown as means ± standard deviation. *P < .05, **P < .01 (Student t test). MFI, mean fluorescence intensity; ns, not significant.
Loss of Twist1 enhances Ca2+ level, mitochondrial activity and metabolism, and ROS level in HSCs upon genotoxic stresses. (A-B) The concentrations of intracellular Ca2+ (A) and mitochondrial Ca2+ (B) in LT-HSCs after IR stress. Representative fluorescence-activated cell sorting (FACS) plots show the fluorescence intensity of Fluo-4 (intracellular Ca2+) and Rhod-2 (mitochondrial Ca2+; n = 5). (C) Mitochondrial membrane potential of LT-HSCs determined by DilC1(5) staining after IR (n = 5). (D) Mitochondrial membrane potential of LT-HSCs determined by TMRM staining after IR (n = 5). (E) Glucose uptake of LT-HSCs after IR (n = 5). (F) Mitochondrial mass of LT-HSCs evaluated by using MTG staining in the presence of verapamil after IR (n = 5). (G) Relative mitochondrial DNA (mtDNA) copy number of LSK cells evaluated by DNA quantitative polymerase chain reaction (PCR; n = 2). (H) Relative ROS levels of LT-HSCs after IR (n = 5). (I) Representative TEM images of mitochondrial morphology in LSK cells after 5-FU treatment (left, 10 000×; right, 20 000×). (J) Expression of Mfn2 messenger RNA based on quantitative reverse transcription PCR and MFN2 protein determined by FACS in LT-HSCs after 5-FU treatment (n = 3-5). (K) Oxygen-consumption rate (OCR) of LSK cells after 5-FU treatment (n = 4). (L) Relative ATP levels of MPPs and LT-HSCs after 5-FU treatment (n = 4). (M) Relative ROS levels of LT-HSCs after 5-FU treatment (n = 5). Data are shown as means ± standard deviation. *P < .05, **P < .01 (Student t test). MFI, mean fluorescence intensity; ns, not significant.
To investigate whether the 5-FU stress–induced defects in Twist1-deficient LT-HSCs were also associated with enhanced transcription of VGCC and mitochondrial function and metabolism, we examined the expression of Cacna1b, Ca2+ level, and mitochondrial properties in LT-HSCs from Twist1 cKO and control mice after 5-FU treatment. We found that Twist1-deleted LT-HSCs exhibited markedly augmented Cacna1b expression, intracellular and mitochondrial Ca2+ levels, ΔΨm, and glucose uptake (supplemental Figure 10A-G). Transmission electron microscopy revealed a fragmented mitochondrial phenotype characterized by several small, round-shaped mitochondria in Twist1-deleted LT-HSCs, suggesting that mitochondrial dynamics were altered (Figure 5I). Therefore, we examined the expression of several genes involved in regulating mitochondrial fusion and fission and found that only the fusion protein mitofusin 2 (MFN2) was significantly altered at both the messenger RNA and protein levels (Figure 5J). To further determine the functional changes in mitochondria, we measured mitochondrial respiration and found a markedly increased oxygen consumption rate (Figure 5K). Furthermore, significantly increased intracellular ATP levels (Figure 5L) and ROS generation (Figure 5M) were observed. Taken together, these results indicate that TWIST1 exerts inhibitory effects on intracellular Ca2+ concentration and mitochondrial function and metabolism in HSCs under both IR and 5-FU stresses.
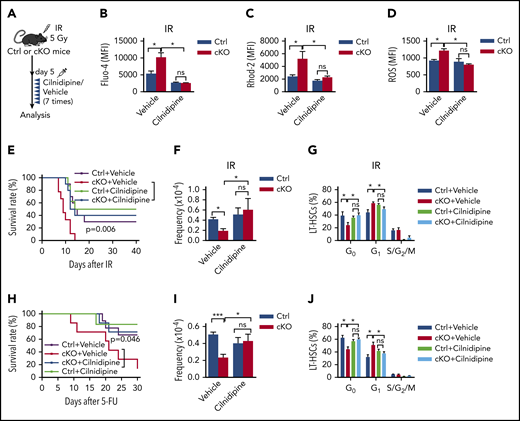
To determine whether the elevated VGCCs mediated the multiple stress-induced defects in Twist1-deleted HSCs, we administered cilnidipine (Figure 6A), a blocker of voltage-gated Ca2+ channels, to irradiated Twist1 cKO and control mice. The results revealed that cilnidipine completely abrogated the increases in intracellular and mitochondrial Ca2+ levels, mitochondrial activity, glucose uptake, and ROS level (Figure 6B-D; supplemental Figure 11A-B). The reduced survival of Twist1-deficient mice was largely prevented (Figure 6E). Additionally, cilnidipine treatment completely rescued HSC frequency and reestablished normal HSC cycling (Figure 6F-G). When Twist1 cKO mice were administered cilnidipine after 5-FU treatment, similar results were observed (Figure 6H; supplemental Figures 11C-F and 12A-C). These findings suggest that TWIST1 preserves the number and function of HSCs by suppressing the CACNA1B/Ca2+/mitochondria pathway under stress conditions.
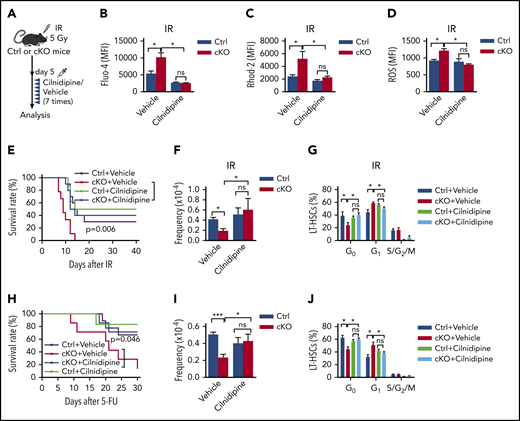
Suppression of VGCC abrogates increased mitochondrial activity and largely rescues HSC defects in Twist1-deficient mice. (A) Schematic overview of the cilnidipine treatment of irradiated mice. Control (Ctrl) and cKO mice were injected with cilnidipine (1 mg/kg per dose; intraperitoneal injection) for 7 consecutive days after IR treatment. (B-D) The concentrations of intracellular Ca2+ (B), mitochondrial Ca2+ (C), and ROS level (D) in BM LT-HSCs from cKO and Ctrl mice treated with cilnidipine or vehicle after IR treatment (n = 4-6; Student t test). (E) Kaplan-Meier survival curves of cKO and Ctrl mice administered cilnidipine or vehicle after IR at 7 Gy (n = 8-10; log-rank test). (F-G) Frequencies (F) and cell-cycle analysis (G) of BM LT-HSCs from cKO and Ctrl mice treated with cilnidipine or vehicle after IR treatment (n = 4-6; Student t test). (H) Kaplan-Meier survival curves of cKO and Ctrl mice administered cilnidipine or vehicle after serial 5-FU injection (n = 6-9; log-rank test). (I-J) Frequencies (I) and cell-cycle analysis (J) of BM LT-HSCs from cKO and Ctrl mice treated with cilnidipine or vehicle (n = 4-5; Student t test). Data are shown as means ± standard deviation. *P < .05, ***P < .001. MFI, mean fluorescence intensity; ns, not significant.
Suppression of VGCC abrogates increased mitochondrial activity and largely rescues HSC defects in Twist1-deficient mice. (A) Schematic overview of the cilnidipine treatment of irradiated mice. Control (Ctrl) and cKO mice were injected with cilnidipine (1 mg/kg per dose; intraperitoneal injection) for 7 consecutive days after IR treatment. (B-D) The concentrations of intracellular Ca2+ (B), mitochondrial Ca2+ (C), and ROS level (D) in BM LT-HSCs from cKO and Ctrl mice treated with cilnidipine or vehicle after IR treatment (n = 4-6; Student t test). (E) Kaplan-Meier survival curves of cKO and Ctrl mice administered cilnidipine or vehicle after IR at 7 Gy (n = 8-10; log-rank test). (F-G) Frequencies (F) and cell-cycle analysis (G) of BM LT-HSCs from cKO and Ctrl mice treated with cilnidipine or vehicle after IR treatment (n = 4-6; Student t test). (H) Kaplan-Meier survival curves of cKO and Ctrl mice administered cilnidipine or vehicle after serial 5-FU injection (n = 6-9; log-rank test). (I-J) Frequencies (I) and cell-cycle analysis (J) of BM LT-HSCs from cKO and Ctrl mice treated with cilnidipine or vehicle (n = 4-5; Student t test). Data are shown as means ± standard deviation. *P < .05, ***P < .001. MFI, mean fluorescence intensity; ns, not significant.
Because TWIST1 directly represses Cacna1b transcription, it is reasonable to speculate that the expression of Cacna1b is increased in Twist1-deleted HSCs and contributes to the defects in HSCs under steady-state conditions. As expected, we observed that the expression of Cacna1b was indeed upregulated in Twist1-deficient HSCs (supplemental Figure 13A), and treatment with cilnidipine not only largely abrogated the reduction in LT-HSCs (Figure 6I) but also reversed the loss of quiescence of these cells (Figure 6J). To further confirm the functional role of CACNA1B in Twist1-deficient HSCs at steady state, we examined the Ca2+ level. As expected, Twist1-depleted LT-HSCs exhibited markedly elevated intracellular Ca2+ and mitochondrial Ca2+ levels, ΔΨm, glucose uptake, ATP levels, and ROS levels (supplemental Figure 13B-H). These data indicate that TWIST1 maintains HSCs via similar mechanisms under both steady-state and stress conditions.
Although OXPHOS engagement is often accompanied by enhanced ROS production, and elevated ROS levels have been identified as key mediators of loss of quiescence and impaired long-term reconstitution capacity, HSC impairment caused by mitochondrial dysfunction is ROS independent in some circumstances.34,37-39 To elucidate whether elevated ROS in Twist1-deficient HSCs upon IR treatment contributes to HSC defects, we treated the animals with the ROS scavenger N-acetyl-l-cysteine (NAC). We found that NAC largely reversed the hypersensitivity of Twist1-deficient mice to high-dose IR and partially rescued the defect in DNA damage of Twist1-deleted HSCs after IR (supplemental Figure 14A-C). In addition, the reduced survival of Twist1-deficient mice upon persistent 5-FU treatment was partly reversed by NAC (supplemental Figure 14D).
Collectively, these data strongly indicate that TWIST1 maintains HSCs by preventing hyperactivation of the CACNA1B/Ca2+/mitochondria axis under steady-state and stress conditions.
Discussion
Here, we demonstrate that TWIST1 is critical for adult HSC maintenance under both steady-state and stress conditions. It functions by repressing the transcription of the VGCC gene Cacna1b, which leads to the subsequent inhibition of the Ca2+/mitochondria pathway. The origin of adult HSCs, a part of the definitive HSC lineage, can be traced back to early embryonic development. Intriguingly, Kulkeaw et al40 recently reported that Twist1 is highly expressed in embryonic HSPCs in the aorta-gonad-mesonephros region and promotes HSPC differentiation. Therefore, TWIST1 is an important transcription factor regulating both embryonic and adult hematopoieses.
Previous studies have reported that although some well-known factors play pivotal roles in regulating HSC resistance to various stresses, KO of their genes in mice has no impact on hematopoiesis under steady-state conditions.41,42 Our present study showed that loss of Twist1 resulted in reduced HSC pool size, impaired quiescence, and myeloid-biased differentiation in steady-state hematopoiesis, suggesting its essential role in HSC regulation. Intriguingly, the reduction of HSCs occurred mainly in lymphoid-biased HSCs, indicating that the enhanced myeloid lineage differentiation in Twist1 cKO mice is probably attributable to the loss of lymphoid-biased HSCs, supporting the notion of a predetermined lineage bias. We also observed that Twist1 deletion led to increased intracellular and mitochondrial Ca2+ concentrations, mitochondrial membrane potential, and glucose uptake in lymphoid-biased, but not in myeloid-biased, HSCs upon IR stress. Furthermore, MFN2, a critical factor in maintaining the pool of lymphoid-biased HSCs,43 was found to be downregulated in Twist1-deficient HSCs after 5-FU treatment. These findings indicate that TWIST1 may be important for the preservation of lymphoid-biased, but not myeloidy-biased, HSCs.
The importance of mitochondrial metabolism, activity, and dynamics in the maintenance of HSC homeostasis has recently been revealed.9,29 Mitochondrial status contributes to the preservation of HSC quiescence and HSC capacity to switch from dormancy to active cell division.44-46 However, the involvement of mitochondria in the modulation of HSCs under stress is poorly understood. Here, we show that TWIST1 protects HSCs from IR- and chemotherapy-induced impairments through the modulation of the Ca2+/mitochondria pathway. Twist1 deletion causes an increase in the Ca2+ level in mitochondria, which in turn leads to enhanced mitochondrial membrane potential and OXPHOS, which ultimately leads to increased ROS and ATP generation. Inhibition of the VGCC reverses the stress-induced alterations in mitochondrial activity, metabolism, and ROS level, leading to rescue of the major defects of HSCs. These data highlight mitochondria as a critical determinant of HSC survival, maintenance, and functional identity under stress. However, it should be pointed out that although the oxygen consumption rate was examined in LSK cells, further investigation is required to determine whether TWIST1 represses mitochondrial respiration in HSCs upon stress.
Calcium is a primary signaling molecule that plays a central role in regulating cell cycle, mitochondrial metabolism, and cell death.47 There have been few studies concerning the role of intracellular Ca2+ in HSC regulation. Our findings demonstrate that low intracellular Ca2+ is crucial for the maintenance of HSCs under both steady-state and stress conditions. Consistent with our conclusions, Luchsinger et al13 recently showed that low Ca2+ suppresses mitochondrial respiration and enhances HSC maintenance in vitro. Umemoto et al14 reported that appropriate suppression of the Ca2+/mitochondria pathway is necessary for HSC self-renewal division in vitro, and increased Ca2+ influx leads to cell-cycle activation of HSCs under stress conditions. However, in the study of Luchsinger et al, low CaCl2 cultures resulted in enhanced HSC cycling, inconsistent with our observation that decreased intracellular Ca2+ contributes to the maintenance of HSC quiescence. A possible explanation for this discrepancy is the difference between in vitro culture conditions and the complicated environment in vivo. HSCs cultured in vitro may have worse maintenance and may more easily enter into active cell cycle compared with those residing in the microenvironment in vivo. Our findings reveal the important regulatory role of Ca2+ in HSC maintenance. Intracellular Ca2+ may well represent a viable target for relieving damaged HSCs and maintaining HSC functions and may provide novel strategies for manipulating HSC cell fate.
Many receptors, channels, calcium-binding proteins, pumps, transporters, enzymes, and transcription factors are involved in the generation and decoding of the different calcium signals in different cells.48 VGCCs mediate the most effective influx of calcium ions into the cell from extracellular space. CACNA1B is highly expressed in the brain and peripheral nervous system and important for regulating neuropathic pain.49 Little is known about its role in HSCs. Here, we observed that Cacna1b was expressed at a low level in HSCs (supplemental Table 2), and TWIST1 promoted the maintenance of HSCs by directly suppressing the expression of Cacna1b. Thus, for the first time, our data reveal that the downregulation of Cacna1b is critical for HSC maintenance.
In summary, this study demonstrates for the first time that TWIST1 plays an important role in maintaining HSC homeostasis by modulating the CACNA1B/Ca2+/mitochondria pathway. Our findings shed new light on the understanding of the molecular basis of HSC fate decisions and contribute to the development of novel avenues to maintain functional HSCs.
RNA-seq and ATAC-seq data have been deposited in the National Center for Biotechnology Information Gene Expression Omnibus database (accession number GSE133682).
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
This work was supported by grants from the National Key Research and Development Program of China (2016YFA0100600), the National Natural Science Foundation of China (82070113, 81730006, and 81421002), and the Chinese Academy of Medical Sciences (CAMS) Innovation Fund for Medical Sciences (2016-I2M-1-017).
Authorship
Contribution: X.M., T.C., and N.W. designed the project and wrote the manuscript; N.W., J.Y., N.Y., S.Y., Y.Z., and Y.R. organized, performed, and analyzed experiments; D.G., H.C., and X.L. performed statistical analysis of the data; Q.R. provided reagents and materials; and all authors approved the final version of the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Xiaotong Ma, State Key Laboratory of Experimental Hematology, National Clinical Research Center for Blood Diseases, Institute of Hematology & Blood Diseases Hospital, 288 Nanjing Rd, Tianjin 300020, China; e-mail: maxt@ihcams.ac.cn; and Tao Cheng, State Key Laboratory of Experimental Hematology, National Clinical Research Center for Blood Diseases, Institute of Hematology & Blood Diseases Hospital, 288 Nanjing Rd, Tianjin 300020, China; e-mail: chengtao@ihcams.ac.cn.