

In this issue of Blood, 1 describe for the first time the occurrence of somatic genetic rescue (SGR) in an asymptomatic individual carrying a pathogenic germline mutation in GATA2. The authors hypothesize that early recovery of hematopoiesis may also have prevented extrahematological features of GATA2 deficiency in this individual. The finding of SGR in GATA2 deficiency stresses once more that awareness for this phenomenon is needed in genetic counseling and testing of pre- or asymptomatic individuals in families with hereditary bone marrow failure.
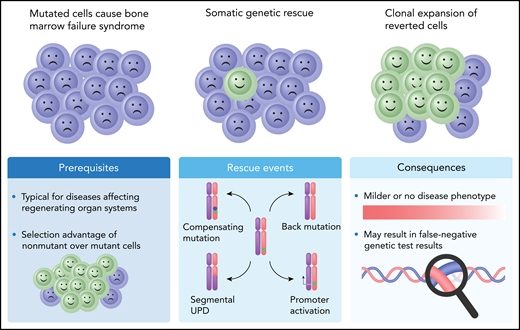
Schematic presentation of SGR in conditions caused by autosomal dominant haploinsufficient genetic variants. Four causative mechanisms for these conditions have been described (Rescue events), either by making use of the functional wild-type allele (reversion by mitotic recombination resulting in segmental UPD, or promoter activation) or by correcting functionality of the mutated copy through a compensating mutation or a back mutation. A compensating mutation (blue dot) occurs on a different location within the same gene copy and takes away the deleterious effect of the pathogenic mutation (green dot), for example, by restoring the reading frame of a pathogenic frameshift mutation. A back mutation reverts the original mutation back to normal. The GATA2 rescue event described by Catto and coworkers is of the “back mutation” type, where in the pathogenic germline GATA2 c.216C>A (p.Y72*) variant, the A residue is spontaneously remutated into a T, by which the amino acid sequence is restored (c.216C>T; p.Y72Y).
Schematic presentation of SGR in conditions caused by autosomal dominant haploinsufficient genetic variants. Four causative mechanisms for these conditions have been described (Rescue events), either by making use of the functional wild-type allele (reversion by mitotic recombination resulting in segmental UPD, or promoter activation) or by correcting functionality of the mutated copy through a compensating mutation or a back mutation. A compensating mutation (blue dot) occurs on a different location within the same gene copy and takes away the deleterious effect of the pathogenic mutation (green dot), for example, by restoring the reading frame of a pathogenic frameshift mutation. A back mutation reverts the original mutation back to normal. The GATA2 rescue event described by Catto and coworkers is of the “back mutation” type, where in the pathogenic germline GATA2 c.216C>A (p.Y72*) variant, the A residue is spontaneously remutated into a T, by which the amino acid sequence is restored (c.216C>T; p.Y72Y).
GATA2 deficiency results from heterozygous loss-of-function mutations in the GATA2 gene, resulting in haploinsufficiency, and is characterized by peripheral blood cytopenias of monocytes, B lymphocytes and natural killer cells, susceptibility to various infections, and a high risk of developing hematologic malignancies, mainly myelodysplastic syndrome and acute myeloid leukemia. Other features are lymphedema, pulmonary alveolar proteinosis, and hearing loss.2 Catto et al report 2 brothers with prominent features of GATA2 deficiency who were shown to carry a nonsense mutation in GATA2. In their asymptomatic father, analysis of leukocyte DNA revealed mosaicism for a silent missense mutation at the exact same codon as the disease-associated GATA2 mutation identified in the sons, with extreme skewing toward the silent mutation. SGR was restricted to the blood lineage, as DNA from sperm and skin fibroblasts still carried the GATA2 nonsense mutation.
With their report, the authors add to a long list of Mendelian hematopoietic diseases in which SGR has been detected.3 SGR is caused by spontaneous correction of a germline pathogenic allele, which results in a selection advantage and clonal expansion of cells lacking the genetic aberration. This phenomenon results in an improvement or even prevention of the hematologic phenotype caused by the germline pathogenic allele.3 In autosomal dominant hematologic disorders associated with haploinsufficiency, the most commonly observed mechanism of SGR is mitotic recombination, resulting in segmental uniparental disomy (UPD) of the chromosome arm that carries the mutated gene and thus replacement of the mutated allele by an extra copy of the wild-type allele. Other, much rarer mechanisms include correction by a back mutation or a compensatory mutation (see figure).1 The SGR mechanism described by Catto and coworkers should be considered a back mutation. Despite the involvement of a second variant, as is observed in compensatory mutations, the rescuing mutation erases the pathogenic variant and reverts the amino acid sequence to wild type (see figure). SGR is one of multiple explanations for variability in phenotypic expression of an inherited mutation. It can be easily overlooked since not all events resulting in genetic rescue are as eye-catching as the compensatory variant described by Catto et al.
Nevertheless, it is still intriguing that this is only the first report of SGR in GATA2 deficiency, whereas large case series of patients with this syndrome have been published.4,5 In hindsight, it may be possible that one or more of the 6 patients with de novo mutations reported by Donadieu et al are children of a parent that experienced SGR.4
Several factors might influence the frequency of SGR in a specific genetic condition, and time is an important one. In GATA2 deficiency, hematologic malignancies are a common first presentation of the disease, often at young age,4 and a genetic rescue thus needs to occur early in life, between conception and malignant hematological clonal expansion. In conditions with a minor susceptibility to leukemia, like dyskeratosis congenita, this timeframe is much larger, creating more opportunity for SGR to occur.6 In the patient described by Catto et al, SGR was present in T cells, which have a long lifespan. This observation made the authors conclude that indeed SGR occurred early in his life.
SGR is driven by a clonal advantage of “rescued cells” above cells carrying the germline mutation. Variability in clonal advantage likely is another factor influencing the frequency of SGR. In individuals with GATA2 deficiency, hematopoiesis in early childhood appears to be grossly intact, and it has been suggested that other genetic, epigenetic, or environmental factors may contribute to bone marrow failure in these patients.7 Possibly SGR can only become apparent in this more severe stage of the disease.
An additional factor of influence on SGR frequency might be the location of the gene relative to the telomeric end of the chromosome. Mitotic recombination, the most frequent event involved, requires the breakpoint to be located centromeric of the mutated gene. Consequently, the more telomeric a gene is located and the longer the chromosome arm, the larger the chance that a mitotic recombination with a break on the centromeric site of the germline mutation will occur. Even multiple events of mitotic recombination can be observed per patient.6,8 For example, TERC, a gene causing dyskeratosis congenita and described to undergo SGR by mitotic recombination, is located toward the telomere of chromosome 3q, whereas GATA2 is located much closer to the centromere of 3q.
Catto et al conclude that their finding suggests that early recovery of hematopoiesis in GATA2 deficiency either by transplant or potentially by gene therapy may be beneficial for hematopoiesis and to prevent other clinical complications. Previous studies have indeed observed a trend that the earlier hematopoietic stem cell transplantation is performed in patients with GATA2 deficiency, the better the outcome.4,9 With the promising development of gene-repair technologies implementing CRISPR/CAS, it is imaginable that patients with hematological conditions amenable for SGR will be no longer dependent on fate for a rescue to occur, but can actively be helped.
Conflict-of-interest disclosure: The authors declare no competing financial interests.

