The molecular consequences of coding mutations can often be predicted simply from their effect on a gene’s sequence. Noncoding mutations require more work. In this issue of Blood, 1 use 3D genomics to make an important contribution to the list of functional noncoding mutations in cancer. They show that microdeletions at 13q12.2 in B-cell precursor acute lymphoblastic leukemia (BCP-ALL) eliminate the boundary of a topologically associated domain (TAD) at the FLT3 locus, which results in higher expression of FLT3, an important driver gene in acute leukemias.
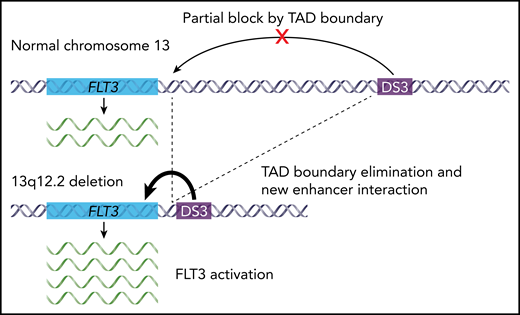
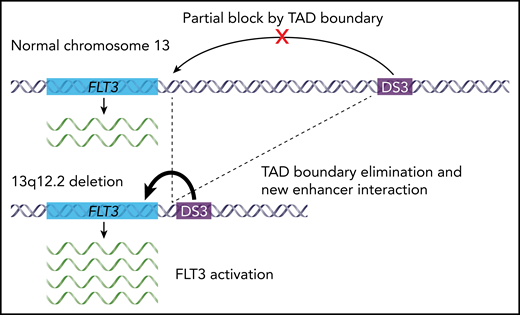
Schematic showing the effect of 13q12.2 deletions on FLT3 expression in BCP-ALL. The novel regulatory enhancer DS3 normally lies far upstream of the FLT3 gene on chromosome 13q. In wild-type cells, DS3 and FLT3 occur in separate TADs and are therefore functionally insulated from one another in the genome. Recurrent 13q12.2 deletions in BCP-ALL eliminate the boundary between these 2 TADs, allowing the DS3 enhancer to contact FLT3 and activate its expression.
Schematic showing the effect of 13q12.2 deletions on FLT3 expression in BCP-ALL. The novel regulatory enhancer DS3 normally lies far upstream of the FLT3 gene on chromosome 13q. In wild-type cells, DS3 and FLT3 occur in separate TADs and are therefore functionally insulated from one another in the genome. Recurrent 13q12.2 deletions in BCP-ALL eliminate the boundary between these 2 TADs, allowing the DS3 enhancer to contact FLT3 and activate its expression.
Altered gene expression is a hallmark of cancer, but the reasons for these changes are often poorly understood. In some cases, somatic mutations directly alter gene expression. For example, chromosomal amplifications and deletions can affect gene expression levels, and some classic translocations reposition oncogenes near highly active promoters or enhancer elements that drive their expression.2,3 The functional consequences of less conspicuous regulatory mutations require more detailed studies. Whole-genome sequencing and chromatin profiling have identified additional regulatory mutations in cancer, including highly recurrent mutations in promoters and distal enhancers.4-6 The ability to measure 3D genome architecture adds another dimension to the search for regulatory mutations in cancer. Connections between gene promoters and noncoding DNA elements in the 3D nucleus help to identify regulatory sequences that are important and the genes that are impacted if these sequences are mutated. Emerging data on the 3D genome have played an important role in the discovery of recurrent regulatory mutations in gastric, lung, and squamous cell cancers.7,8
Yang et al used information about the 3D genome to investigate the functional consequences of a recurrent heterozygous deletion in BCP-ALL patients. These deletions occur at 13q12.2 near FLT3, a gene with clear biological and clinical relevance in myeloid and lymphoid leukemias. The authors previously identified 13q12.2 deletions by whole-genome sequencing,9 and in this study, they combined data from 1418 BCP-ALL patients to characterize deletion frequency, genetics, and associations with clinical and molecular features. Although deletions at 13q12.2 were found in less than 2% of all cases, the authors showed that they are associated with high hyperdiploid BCP-ALL, and they used the set of deleted alleles to define the commonly deleted region. Given its recurrence and proximity to FLT3, they then hypothesized that the deletions may affect FLT3 regulation. Indeed, analysis of RNA-sequencing data from patient samples showed that cases with the deletion had higher FLT3 expression. These expression changes were relatively modest, but analysis of linked single nucleotide polymorphisms at the locus showed skewing of the expressed FLT3 alleles, with higher expression of alleles in cis with a deletion. Data on epigenetic modifications and chromatin interactions added the final mechanistic details. Analysis of histone modifications identified multiple DNA elements in the region that were consistent with regulatory enhancers. One of these enhancers occurred far beyond the deleted region, which 3D genome studies of wild-type lymphocytes showed is normally in a separate TAD, and therefore functionally insulated from FLT3. However, in situ HiC data from a primary BCP-ALL sample with a 13q12.2 deletion showed that this TAD boundary was weakened, and new cis interactions formed between the FLT3 promoter and the novel distal enhancer (see figure).
Together these data provide compelling evidence that recurrent 13q12.2 deletions are directly responsible for increased FLT3 expression in these patients, which is a novel mechanism of FLT3 activation in BCP-ALL. This result is significant for at least 3 reasons. First, it contributes valuable functional annotation to the 13q12.2 region regarding the role of the TAD boundary affected by the deletion and the functional properties of the distal enhancer that hijacks FLT3 in deleted cells. These annotations advance our general understanding of the human genome and how this specific locus is regulated in hematopoietic cells. Second, the availability of therapeutic FLT3 inhibitors10 means that 13q12.2 deletions could have clinical implications for patients with BCP-ALL and perhaps for other patients with acute leukemia. Although the study by Yang et al is too small to be definitive, 13q12.2 deletions seem to be stable through relapse. This could mean that they are driver mutations, and if BCP-ALL cells with 13q12.2 deletions are genetically addicted to FLT3 signaling, then treatment with anti-FLT3 therapies could be effective. FLT3 is also an important gene in acute myeloid leukemia, and the findings here raise the possibility that 13q12.2 deletions could occur in these patients and explain some of the variability in FLT3 expression in this malignancy. The detailed analysis presented in this article offers a guide for detecting 13q12.2 deletions and their molecular consequences in genome sequences and gene expression data.
Third, the study by Yang et al adds to the list of recurrent mutations in cancer that act by disrupting normal gene regulation. Compared with coding mutations, this list is relatively short, and clear examples of the typical genomic features, functional consequences, and approaches required to characterize these mutations are needed. For example, this study demonstrated that 13q12.2 deletions were associated with relatively subtle increases in FLT3 expression, which ultimately required integration of multiple genomic data sets to arrive at a final mechanistic explanation. This is what it will take to uncover additional mutations like the 13q12.2 deletion. They are certainly out there. It just takes more work to find them.
Conflict-of-interest disclosure: The author declares no competing financial interests.