Abstract
Pyruvate kinase deficiency (PKD) is an autosomal-recessive enzyme defect of the glycolytic pathway that causes congenital nonspherocytic hemolytic anemia. The diagnosis and management of patients with PKD can be challenging due to difficulties in the diagnostic evaluation and the heterogeneity of clinical manifestations, ranging from fetal hydrops and symptomatic anemia requiring lifelong transfusions to fully compensated hemolysis. Current treatment approaches are supportive and include transfusions, splenectomy, and chelation. Complications, including iron overload, bilirubin gallstones, extramedullary hematopoiesis, pulmonary hypertension, and thrombosis, are related to the chronic hemolytic anemia and its current management and can occur at any age. Disease-modifying therapies in clinical development may decrease symptoms and findings associated with chronic hemolysis and avoid the complications associated with current treatment approaches. As these disease-directed therapies are approved for clinical use, clinicians will need to define the types of symptoms and findings that determine the optimal patients and timing for initiating these therapies. In this article, we highlight disease manifestations, monitoring approaches, strategies for managing complications, and novel therapies in development.
Introduction
Pyruvate kinase deficiency (PKD), the most common defect in the glycolytic pathway associated with congenital hemolytic anemia, was first described in the early 1960s. After Selwyn and Dacie1 initially discovered the link between glycolysis and hemolytic anemia in the 1950s, De Gruchy and colleagues2 reported a series of patients with nonspherocytic hemolytic anemia whose hemolysis could be corrected by adenosine triphosphate (ATP), but not by glucose. Soon thereafter, the molecular basis of this anemia was described by Valentine3 and Tanaka4 and their colleagues as being due to pyruvate kinase (PK) deficiency.
Healthy red blood cells (RBCs) rely mainly on glycolysis for generation of red cell ATP. PK catalyzes the conversion of phosphoenolpyruvate to pyruvate, resulting in ATP production. ATP is essential for maintaining the structural and functional integrity of RBCs during their lifespan of 100 to 120 days. Abnormal or deficient pyruvate kinase (PK) enzyme activity results in inadequate ATP production, with consequent loss of membrane plasticity, cellular dehydration, and premature destruction of RBCs in the spleen or liver.5-8 Reticulocytes, which require high levels of ATP, are particularly susceptible to dehydration and injury in the hypoxic spleen as a result of their shift from using oxidative phosphorylation to glycolysis.7,8 Deficient PK activity also results in the accumulation of 2,3-diphosphoglycerate (2,3-DPG), an upstream product of the glycolytic pathway that causes a rightward shift in the hemoglobin-oxygen dissociation curve.9,10
PKD is caused by autosomal-recessive variants in the PKLR gene located on chromosome 1q21. The genetic control of PK explains why the disease findings are confined to red cells. Two genes, PKLR and PKM, encode the 4 PK isoenzymes. The M1 and M2 PK isozymes are encoded by PKM on chromosome 15. PK-M2 is the major isozyme of erythroid precursors. The PK-L (liver) and PK-R (RBC) isozymes are homotetramers encoded by PKLR. During normal erythroid differentiation, the PK isozyme switches from PK-M2 to PK-R. Because hepatocytes retain the capacity for protein synthesis and have residual PK-M2 activity, the liver is generally unaffected in PKD, whereas mature red cells are dependent on glycolysis and rely exclusively on PK-R.
PKD is highly heterogeneous from biochemical and genetic points of view, because homozygotes generally exhibit <25% residual RBC enzyme activity in vitro, whereas heterozygotes have 40% to 60% activity; >300 pathogenic mutations in the PKLR gene have been described.11 Since the initial description, cases of PKD have been reported worldwide.12-19
Epidemiology
The frequency of PKD is not precisely defined, but it has an estimated prevalence of 3 to 8 per 1 000 000.20-23 Given its rarity, challenges in the diagnosis, and the broad spectrum of clinical symptoms, many cases may be undiagnosed; therefore, this frequency may be an underestimate. Heterozygous carriers are typically asymptomatic, which makes the prevalence difficult to determine, but it likely ranges from 0.15% to 6%. Patient communities with higher frequencies of carriers and cases of PKD are due to a founder effect.21,24 PKD has a wide geographic distribution and may have an advantage with regard to selective pressure in certain areas of the globe due to protection from malaria.25-27
Diagnosis
The diagnosis of PKD should be suspected in the presence of clinical signs and symptoms and laboratory markers of chronic hemolytic anemia, including splenomegaly, jaundice, gallstones, increased reticulocytes, indirect hyperbilirubinemia, and mild hyperferritinemia. Clinical presentation may be different by age groups and is discussed in the sections on newborns, children, and adults. Because PKD is an autosomal-recessive disorder, the family history is typically unrevealing, with the exception of miscarriages and affected siblings. The differential diagnosis includes the heterogeneous group of congenital and acquired hemolytic disorders.11,28 The diagnosis is based on the exclusion of more common causes of hemolysis, on the demonstration of reduced PK enzymatic activity, and on the detection of compound heterozygous and homozygous mutations in the PKLR gene.11
PK enzymatic activity is usually determined in RBC lysates by spectrophotometric assay. Recent transfusions or incomplete removal of platelets or white blood cells may give falsely normal levels. In addition, PK enzyme activity is red cell age–dependent, with the highest levels in reticulocytes, which can lead to falsely normal PK levels in some affected patients due to reticulocytosis. The diagnosis should be suspected in patients with a normal PK activity level that is relatively low in comparison with other red cell age–dependent enzymes, such as hexokinase. Notably, there is no clear correlation between the degree of in vitro enzyme activity and clinical severity.13,29 Decreased PK activity may also be found in heterozygous carriers.11 Therefore, the diagnosis of PKD should be confirmed by PKLR genotyping. The majority of PKLR mutations are missense substitutions. Common missense variants include R510Q in northern Europe and the United States, R486W in southern Europe, and the R479H mutation in Amish communities.30,31 Disruptive mutations, such as stop codons and large deletions, are less frequent and are generally associated with a more severe phenotype.29 Most patients are compound heterozygotes, which, in addition to the large number of reported PKLR variants, makes studies of genotype-phenotype correlations challenging. Affected patients in the Amish community are homozygous for the R497H variant but have wide clinical variability, despite genotypic homogeneity.29,30 Genetic testing requires a small volume, is not affected by transfused RBCs, and is suitable for prenatal diagnosis. However, large deletions and intronic mutations can be difficult to identify, and further studies should be pursued in cases with high clinical suspicion but negative genetic testing. In addition, newly reported mutations, in ∼20% of currently diagnosed patients, should be classified as causative by functional tests. Therefore, both PK enzyme activity and PKLR genetic testing are recommended to confirm the diagnosis of PKD.11,28
Clinical presentation and complications
The clinical presentation of PKD is variable and ranges from in utero complications, to incidentally noted indirect hyperbilirubinemia or reticulocytosis associated with fully compensated hemolysis without anemia, to symptomatic anemia leading to regular blood transfusions. Patients most often present within the first month of life; however, diagnoses in adulthood are not uncommon, particularly in patients with compensated hemolysis or mild anemia or who were misdiagnosed with another hemolytic disorder.
Symptoms and management in newborns
In utero complications of affected infants include hydrops fetalis, intrauterine growth retardation, and prematurity.32,33 Affected pregnancies may require in utero blood transfusions. Rarely, newborns can present critically ill at the time of birth, with evidence of pulmonary hypertension and/or stroke. Extramedullary hematopoiesis of the skin may be evident at birth. Liver failure is a rare presentation in newborns with PKD, likely related to a deficiency in both the PK-L and PK-M2 isozymes leading to ATP deficiency in the hepatocytes; it is associated with a transaminitis and direct hyperbilirubinemia and can progress to hepatocellular injury and synthetic dysfunction.34,35 These patients have a high mortality, but several cases have been successfully managed with liver transplant.36
Neonatal jaundice is common, with many newborns experiencing severe indirect hyperbilirubinemia requiring phototherapy.21,29,30 Exchange transfusions may also be required to reduce the risk of kernicterus and historically have been necessary in 46% of births.29 If the hemolysis is recognized early, aggressive phototherapy and hydration may help to avoid an exchange transfusion. Upon birth, infants are often found to have hemolysis requiring transfusions. The physiologic nadir can be prolonged in transfused infants, making it difficult to assess the baseline hemoglobin nadir and ongoing transfusion needs. However, newborns may also have no evidence of jaundice and/or only mild anemia; in these cases, PKD may not be diagnosed until a deeper symptomatic physiologic hemoglobin nadir is uncovered in the first 4 to 8 weeks of life or with evidence of hemolysis even later in childhood or adulthood.
Symptoms and supportive management in children
Laboratory findings and symptoms in children
In nontransfused nonsplenectomized children, the median hemoglobin value is 9 g/dL (range, 6-12.5; Table 1). Splenectomy partially ameliorates the hemolytic anemia and decreases the transfusion burden in 90% of patients and raises the hemoglobin a median of 1.6 g/dL.29 A paradoxical increase in reticulocyte count is seen after splenectomy. A modest elevation in lactate dehydrogenase may be seen, particularly with episodes of increased hemolysis, but it is commonly in the normal range (Table 1). Splenomegaly is common (80-85%), with mild hypersplenism seen in some patients with associated mild leukopenia and thrombocytopenia and/or increased transfusion needs.29
Children have an indirect hyperbilirubinemia (bilirubin range, 2-6 mg/dL) that does not improve after splenectomy. Children with concurrent Gilbert syndrome will have higher indirect bilirubin levels. Jaundice and scleral icterus are almost universal in children with PKD (40-70%), particularly for patients with associated Gilbert syndrome, and they can lead to substantial social impacts.29
The symptoms associated with anemia in young children and adolescents are variable, and patients with the same hemoglobin value can have significantly different symptoms and findings. The low hemoglobin may be better tolerated by patients with PKD because of the elevation of 2,3-DPG, in comparison with other forms of anemia. An early study demonstrated increased oxygen extraction with exercise in a patient with PKD compared with a patient with hexokinase deficiency.37 Examination of actual exercise tolerance in individuals with PKD and other anemias deserves further study, but differences in red cell 2,3-DPG between patients with PKD may partially explain the variability in symptoms among patients, despite similarly low hemoglobin levels.
Infants and children with low hemoglobin values may have poor feeding, growth, and energy levels and develop irritability. Young children can develop frontal bossing related to increased intramedullary erythropoiesis. Older children and adolescents may have poor school focus, persistence of naps, and low energy to participate in activities. However, some patients with PKD do not have fatigue, despite a low hemoglobin. Given this, it is important to assess symptoms carefully when determining patient management and need for interventions, including transfusions and splenectomy.
Transfusions in children
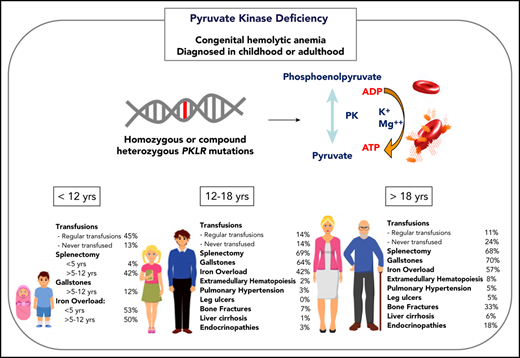
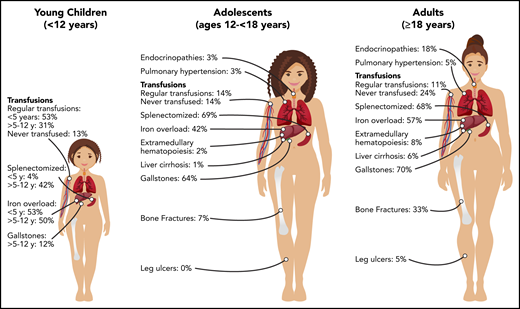
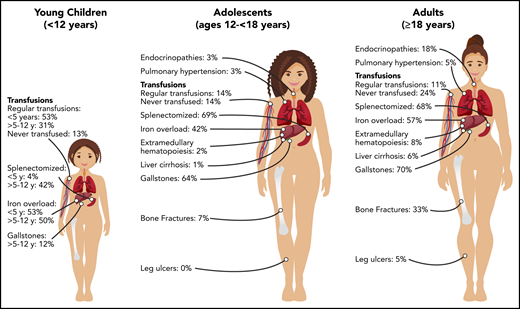
Transfusions in children with PKD are common, with 87% of patients younger than 18 years of age having received ≥1 transfusion in their lifetime (Figure 1). Intermittent transfusions are often needed in the setting of increased hemolysis from acute stressors, including viral infections. For some children, recurrent viral infections in early childhood may give the appearance of transfusion dependence because of frequent episodes of increased hemolysis.
Burden of morbidities, management, and complications in young children (<12 years), adolescents (ages 12 to ≤18 years), and adults (≥18 years) with PKD.
Burden of morbidities, management, and complications in young children (<12 years), adolescents (ages 12 to ≤18 years), and adults (≥18 years) with PKD.
The decision to transfuse at regular intervals relates to a number of factors, including the patient’s growth, daily symptoms (including level of activity and impact of anemia on quality of life), complications, and, to a lesser extent, the nadir hemoglobin level (Figure 2). The threshold and interval between transfusions is set by the goal of avoiding a hemoglobin nadir at which a patient develops symptoms. For children younger than 5 years of age, ∼50% are regularly transfused at an average interval of 5 weeks. Most of these patients will have a splenectomy around the age of 5 years and then will not be transfused or will only need transfusions for acute stressors. Transfusions become less frequent throughout childhood related to the timing of splenectomy and the frequency of viral infections, with regular transfusions in only 30% of children ages 5 to 12 years and 14% of those ages 12 to 18 years.29 Allosensitization is rare in PKD, reported in only 2% of patients.
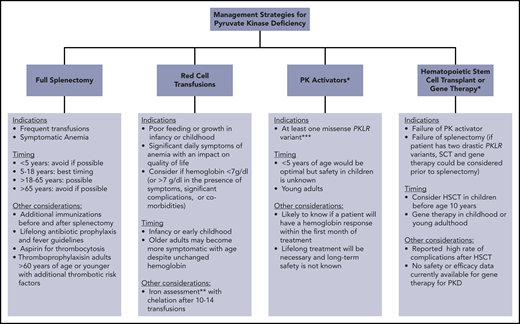
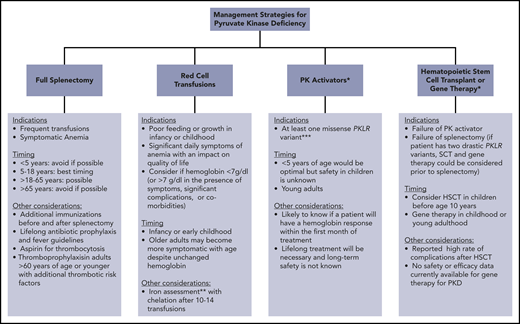
Clinically available and research-based management strategies for patients with PKD. *Research treatment in clinical development. **Iron overload often occurs in the absence of transfusion. Chelation therapy should be strongly considered if liver iron concentration > 5 mg/g or ferritin > 800 μg/L and transferrin saturation > 60%, even in the absence of transfusions. ***Future studies may indicate that other genotypes are responsive.
Clinically available and research-based management strategies for patients with PKD. *Research treatment in clinical development. **Iron overload often occurs in the absence of transfusion. Chelation therapy should be strongly considered if liver iron concentration > 5 mg/g or ferritin > 800 μg/L and transferrin saturation > 60%, even in the absence of transfusions. ***Future studies may indicate that other genotypes are responsive.
As in other hemolytic anemias, patients with PKD will develop an aplastic crisis, with profound anemia and reticulocytopenia, in the setting of parvovirus infection; this occurs most frequently in childhood and only once in a lifetime. Occasionally, this may be the initial manifestation of PKD.
Splenectomy in children
Splenectomy is recommended in children who receive frequent transfusions, and it can also be beneficial in those with daily symptoms related to chronic anemia (Figure 2).38 However, compared with many other congenital hemolytic anemias, splenectomy only partially improves the anemia in PKD and is not effective for all patients.8,39-42
Splenectomy is typically delayed until after the age of 5 years to decrease the risk of sepsis due to encapsulated organism bacteremia. The timing is based on a balance between the risks of postsplenectomy sepsis vs the risks of red cell transfusions and iron loading. The decision to proceed with splenectomy is variable, based on physician practice and patient preferences. Approximately 5% of patients will undergo splenectomy before age 5 years. By age 12 years, 40% will have had a splenectomy and, by age 18 years, 70% will be splenectomized. Splenic studies, including measuring spleen size or red cell survival, do not predict which patients will have a hemoglobin response to splenectomy, because much of the hemolysis occurs in the liver as well as in the spleen. Patients with the most severe hemolysis are least likely to have a significant increase in hemoglobin after splenectomy, including those with a lower presplenectomy hemoglobin, 2 drastic PKLR mutations, and a higher total bilirubin level.29 After splenectomy, hemolysis persists, correlating with a continued risk for pigmented gallstones and their complications.
The decision if and when to proceed with splenectomy needs to be considered carefully. Regardless of vaccine status and/or prophylactic antibiotics, splenectomy increases the susceptibility to serious bacterial infections with encapsulated organisms and other organisms, such as malaria and babesia.43
Iron overload in children
Iron overload is seen at all ages and in regularly transfused and nontransfused or infrequently transfused patients with PKD. Approximately 50% of children younger than 18 years of age have iron overload. The risk of iron loading is lifelong and does not change by age (Figure 1; Table 1).48
After 10 to 14 red cell transfusions, iron overload has occurred and can lead to toxic circulating free iron. For children requiring regular transfusions, this typically occurs between the ages of 1 and 2 years. In these children, magnetic resonance imaging (MRI) for iron measurement, if available, should be performed and followed on an annual basis while patients are chelated.
Increased intestinal iron absorption related to chronic anemia and ineffective erythropoiesis leads to nontransfusion-related iron loading.48,49 In children who have received fewer than 10 to 14 transfusions, annual monitoring of ferritin is recommended. If MRI assessment of iron overload is available, nonregularly transfused patients should have an MRI once the patient is at an age at which the MRI can be conducted without sedation and/or has a ferritin level > 500 μg/L (Table 2).
The approach to chelation in PKD is similar to other red cell and iron loading disorders.50,51 In nontransfused patients, therapeutic phlebotomy can be considered, but it is often not tolerated in significantly anemic or symptomatic patients.
Gallstones/cholecystectomy in children
Gallstones occur in ∼20% of children with PKD, with a median age at diagnosis of 15 years.29 Risk factors for gallstones include a higher indirect bilirubin level from a high hemolytic rate and/or coinherited Gilbert syndrome. Hemolytic rates remain high even after splenectomy; therefore, cholecystectomy should be considered at the time of splenectomy, even in those without gallbladder findings, to avoid future complications and/or a second surgery. However, patients should be counseled that intrahepatic cholestasis can occur even after cholecystectomy.
Symptoms and supportive management in adults
Laboratory findings and symptoms in adults
In nontransfused nonsplenectomized adults, the hemoglobin levels range from 7.6 to 14.2 g/dL (Table 1), with a higher median hemoglobin reported compared with children with PKD (11.3 vs 9.0 g/dL), likely related to the timing of splenectomy. Similar to children, jaundice, scleral icterus, and splenomegaly are frequent findings in adults. The clinical variability of the disease is also evident in adults with some patients exhibiting few symptoms of anemia and others with increasing symptoms with age and additional comorbidities despite a stable hemoglobin level. Reports of fatigue, shortness of breath, memory loss, difficulty concentrating, and bone pain are common.52 Adults may have difficulty participating in responsibilities outside of work or in maintaining full-time employment because of fatigue. Given the variability in symptoms in adults with PKD, it is important to regularly assess symptoms carefully to determine individual management.
Transfusions, splenectomy, and complications in adults
The frequency of patients who receive regular transfusions decreases with age (11% of adults vs 36% of children), which is related to less frequent hemolytic episodes associated with infections as well as the timing of splenectomy (Figure 2). However, the symptoms of anemia may increase with age and comorbidities, and some adults may reinitiate regular transfusions to decrease symptoms of anemia despite being nontransfused for many years. The decision to transfuse is based on the impact of anemia on daily quality of life and the complications associated with chronic hemolysis (Figure 2).
Splenectomy is also considered in adults with PKD. Although the optimal timing is likely between the ages of 5 and 18 years, splenectomy can be considered through adulthood in those receiving frequent transfusions or with daily symptoms from anemia (Figure 2). The infectious and thrombotic risks persist lifelong after splenectomy; thus, vigilance is required with regard to optimizing vaccines, prophylactic antibiotics, fever management, and thromboprophylaxis.
Iron overload is common in adults who are regularly transfused and in those who are nontransfused or infrequently transfused. This is an underrecognized complication in nontransfused adults and requires regular monitoring (Table 2). The approach to chelation in adults with PKD is similar to the approach in children.
Because the disease has a chronic course, the rate and number of complications, with the exception of iron overload, are typically greater in adults than in children (Figure 1). Gallstones have affected 70% of adults compared with 21% of children, and endocrine dysfunction, including thyroid disease, hypoparathyroidism, diabetes, and hypogonadal hypogonadism, is also observed more frequently in adults (18% in adults vs 3% in children). Leg ulcers, which have not been reported in children, are observed in 5% of adults.53 Osteopenia and bone fractures may be observed in up to 33% of adults. The rate of extramedullary hematopoiesis is greater in adults (8% vs 2%), likely reflecting chronic marrow compensation.54-56 Pulmonary hypertension is reported in 5% of adults and can be associated with significant morbidity.57 Reports of depression and anxiety are not uncommon as a result of accumulating symptoms and complications; clinicians should monitor for these symptoms and recommend psychological support when indicated.
Management of PKD in pregnancy
During pregnancy, the degree of hemolysis typically worsens, and transfusion needs increase significantly.30,58-60 Most women will be transfused during the pregnancy or after the delivery. Transfusions should be strongly considered to support a higher hemoglobin nadir during pregnancy to allow for normal fetal growth. Multidisciplinary care with a hematologist and high-risk obstetrician is recommended with close monitoring of fetal growth.59 Preterm births occur in ∼10% of pregnancies of affected mothers, and 18% of pregnancies result in a miscarriage.29
In addition to monitoring the growth of the unaffected fetus, pregnant women should be screened for hepatitis B and C and HIV if they have ever been transfused. An echocardiogram could also be considered prior to or around the time of conception to evaluate cardiac function and the presence of pulmonary hypertension. Folic acid supplementation should be started prior to conception. Prenatal vitamins containing iron during pregnancy should be avoided unless there is evidence of iron deficiency through laboratory iron testing.
Disease-modifying treatments
PK activators
Several activators of red cell PK are in clinical development. Mitapivat, AG-348, is an allosteric small molecule PK activator that has been shown to activate wild-type and a wide spectrum of mutated PK enzymes in vitro.61 In patient samples ex vivo and in PKD mouse models, mitapivat enhances glycolytic flux.61 In a phase 1 study of healthy individuals, mitapivat induced PK activity in vivo, increased red cell ATP, and decreased 2,3-DPG.62 Following this study, a global phase 2 trial enrolled 52 adults with PKD, with a median hemoglobin of 8.6 g/dL, who had not received transfusions in the prior 4 months.63 A hemoglobin increase > 1.0 g/dL was seen in 50% of the participants, with a mean hemoglobin increase of 3.4 g/dL (range, 1.1-5.8). Improvement in markers of hemolysis correlated with the increase in hemoglobin. Hemoglobin response occurred over a median of 10 days and was independent of splenectomy status. Hemoglobin response was sustained with up to 35 months of ongoing treatment. A relationship between genotype and hemoglobin response was observed: patients with at least 1 missense mutation had a higher likelihood of a hemoglobin response. None of the patients with 2 drastic mutations or from the Amish community (homozygous R479H variant) had a hemoglobin response. In addition, a relationship between red cell PK protein level and hemoglobin response was observed, likely indicating that a minimum amount of full-length PK protein is required for activation by mitapivat.
The most common adverse events with mitapivat were headache, insomnia, and nausea, which occurred most often within the first week of drug initiation and were transient. Mitapivat has an off-target effect as an aromatase inhibitor. Sex hormone changes primarily remained within normal ranges, did not correlate with changes in bone mineral density, and were reversible upon drug discontinuation. Patients in whom the drug was held because of an early robust hemoglobin response were found to have withdrawal hemolysis, indicating that patients with a hemoglobin response require a dose taper when coming off mitapivat. Studies of safety and efficacy in children have not been conducted. The currently enrolling phase 3 trials have an individualized dose-escalation schema to minimize risks and achieve efficacy (NCT03559699, NCT03548220).
Because the majority of patients with PKD are compound heterozygotes with at least 1 missense PKLR mutation, mitapivat has the potential to increase hemoglobin levels in most patients. Given this, if mitapivat or another PK activator becomes available on a clinical basis, it should be considered in patients with PKD of all ages, particularly those with at least 1 missense mutation (Figure 2).
Stem cell transplant
The indications for hematopoietic stem cell transplant (HSCT) in patients with PKD are not well-defined (Figure 2). Although HSCT has the potential to cure PKD, current approaches are associated with a relatively high rate of morbidity and mortality compared with standard supportive care. Animal models have shown that HSCT can successfully correct hemolytic anemia associated with PKD.64 Between 1996 and 2015, 16 patients with PKD were transplanted, all from Europe and Asia.65 In this cohort, the median age at transplantation was 6.5 years, with a median follow-up of 2.3 years. Grade 3-4 graft-versus-host disease was reported in 7 of 16 patients, with a 2-year cumulative survival of only 74%. In this cohort, 5 of 16 patients died from transplant-related causes. These patients had varying donor types, conditioning regimens, and graft-versus-host disease and infection prophylaxis. A significantly better survival was observed in patients transplanted before 10 years of age, suggesting that transplant is best considered in the first decade of life. Despite the reported success of HSCT in several patients, given the available data regarding the risks for transplant and the long-term course of PKD, splenectomy and/or regular red cell transfusions are generally recommended rather than HSCT. HSCT could be considered in young patients with 2 drastic PKLR mutations who are regularly transfused despite splenectomy. If a patient is eligible to trial a PK activator, strong consideration should be made for trialing this first and considering transplant in those who fail to respond or tolerate this treatment. Further study of the role and safety of HSCT in PKD is needed.
Gene therapy
As a single-gene defect primarily affecting RBCs, gene therapy is a promising option for PKD. Published studies have been limited to animal models of PKD.66-70 A lentiviral vector was used to transduce mouse PKD hematopoietic stem cells that were subsequently transplanted into myeloablated mice with a PKD phenotype. The procedure normalized the erythroid compartment, correcting the hematologic phenotype and reverting organ pathology with normalization of the spleen size.66 Metabolic studies demonstrated functional correction of the glycolytic pathway with no metabolic disturbances in leukocytes and no evidence of genotoxicity. These promising preclinical results support future human gene therapy for PKD. A currently enrolling phase 1 clinical trial of gene therapy using a lentiviral vector will assess the safety in adults and children with PKD who have hemoglobin < 9.5 g/dL despite splenectomy and receive frequent transfusions (NCT04105166). In the future, if available on a clinical basis, the indications for gene therapy will need to be considered. If effective, gene therapy could be considered in young patients with 2 drastic PKLR mutations who are regularly transfused despite splenectomy (Figure 2). If a patient is eligible to trial a PK activator, strong consideration should be made for trialing this first and considering gene therapy in those who fail to respond or tolerate this treatment.
Conclusions
PKD is a lifelong chronic hemolytic anemia with a wide spectrum of symptoms and manifestations. Given the significant risk of complications that can arise over a patient’s lifetime, monitoring is critical. Currently, supportive care includes transfusions, splenectomy, and chelation therapy. PK activators and gene therapy offer innovative disease-directed approaches that may transform the clinical phenotype of patients in the future. Given the potential future treatment possibilities for PKD, careful thought is needed to determine the optimal management strategies in individual patients.
Authorship
Contribution: R.F.G. and W.B. cowrote the manuscript.
Conflict-of-interest disclosure: W.B. receives consulting and research funding from Agios Pharmaceuticals. R.F.G. is a scientific advisor and consultant for, and receives research funding from, Agios Pharmaceuticals.
Correspondence: Rachael F. Grace, Dana-Farber/Boston Children’s Cancer and Blood Disorders Center, 450 Brookline Ave, D3-106, Boston, MA 02215; e-mail: rachael.grace@childrens.harvard.edu.