Abstract
Although allogeneic hematopoietic cell transplantation (allo-HCT) is currently the standard curative treatment of acute leukemia, relapse remains unacceptably high. Measurable (minimal) residual disease (MRD) after allo-HCT may be used as a predictor of impending relapse and should be part of routine follow-up for transplanted patients. Patients with MRD may respond to therapies aiming to unleash or enhance the graft-versus-leukemia effect. However, evidence-based recommendations on how to best implement MRD testing and MRD-directed therapy after allo-HCT are lacking. Here, I describe our institutional approach to MRD monitoring for preemptive MRD-triggered intervention, using patient scenarios to illustrate the discussion.
Introduction
While >30 000 allogeneic hematopoietic cell transplantations (allo-HCTs) are performed annually worldwide, some 30% of the recipients are destined to relapse within 2 years of allo-HCT.1-3 Measurable (previously termed minimal) residual disease (MRD) monitoring in acute myeloid leukemia (AML) and acute lymphoblastic leukemia (ALL) has been introduced into clinical practice as a validated tool for early prediction of subsequent relapse and to improve outcomes by guiding subsequent therapy, including decisions regarding transplantation.4-6 MRD persistence at transplant has been identified as the strongest risk factor for posttransplant relapse,7,8 which may be at least partially overcome by additional intervention such as blinatumomab in B-cell ALL or augmented conditioning in AML.9,10 Still, there is a relative paucity of data regarding MRD and MRD-driven interventions following allo-HCT. MRD after transplant is associated with an increased incidence of relapse, but the clinical implications of MRD kinetics are not yet clearly defined. All the methodological considerations and limitations that complicate MRD testing in the nontransplant setting, such as a lack of leukemia-specific markers, nonstandardized techniques, poorly defined cutoffs and testing intervals, and MRD-target loss due to clonal/molecular evolution, also beleaguer posttransplant MRD monitoring.4 Furthermore, the clinical interpretation of MRD after allo-HCT is even more enigmatic (Table 1). Unlike chemotherapy, which induces an antileukemic effect of short duration, the GVL effect is prolonged, with unique and nonquantifiable dynamics in different individuals, and may require several months to eradicate any persisting tumor cells.11 The strong and protracted pressure of GVL may promote immune evasion in persisting leukemia cells, such as through genomic loss or downregulation of HLA genes, immune-checkpoint ligand overexpression, or generation of an anti-inflammatory milieu in the leukemic microenvironment, resulting in resistance to GVL.12-14 Patients with MRD may respond to discontinuation of immunosuppression and donor lymphocyte infusion (DLI), but convincing evidence that preemptive intervention strategies will improve outcome is lacking (Table 2).
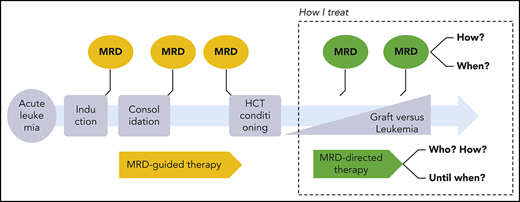
How should we monitor MRD in acute leukemia after allo-HCT, when should we react, and how should we intervene? Published workshop guidelines,15-17 expert opinions,18,19 and consensus statements20 differ from evidence-based facts.21 The cases below illustrate our individualized, risk-adapted, but still somewhat intuitive approach in everyday clinical practice (Figure 1).
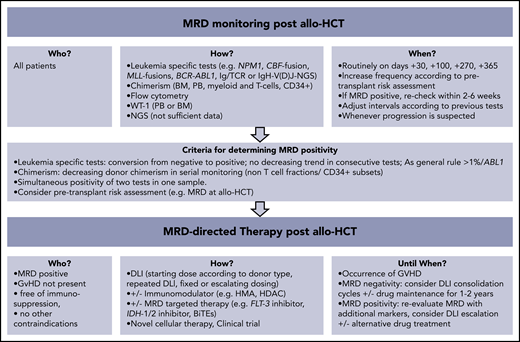
How I monitor and treat MRD after allo-HCT. BiTEs, bi-specific T-cell engagers; HDAC, histone deacetylase; HMA, hypomethylating agents.
How I monitor and treat MRD after allo-HCT. BiTEs, bi-specific T-cell engagers; HDAC, histone deacetylase; HMA, hypomethylating agents.
How do I monitor MRD after transplantation, and when do I react?
Case 1
A 56-year-old woman with relapsed NPM1mut AML underwent myeloablative conditioning (MAC) allogeneic peripheral blood stem cell transplantation (PBSCT) from an 9/10 HLA-matched unrelated donor in her second complete remission (CR). Pretransplant bone marrow (BM) sampling showed 170 (1.7%) NPM1mut/104ABL1 copies by real-time quantitative polymerase chain reaction (RT-qPCR). Graft-versus-host disease (GVHD) prophylaxis consisted of low-dose alemtuzumab (10 mg) and cyclosporine, which was discontinued at day +122.22 At 3 months after allo-HCT, BM-MRD was positive with 45 NPM1mut/104ABL1 copies while 4-color FC was negative for leukemic blasts (sensitivity 1%). Repeated analyses 4 and 7 weeks after initial testing showed persistent BM-MRD (55 and 97 NPM1mut/104ABL1 copies, respectively). Chimerism evaluation with short tandem repeat (STR)-based PCR (sensitivity 1% to 5%) depicted complete donor chimerism (CC) in both BM and peripheral blood (PB) at all times.
NPM1mut monitoring
Ideally, pretransplant NPM1mut MRD-positive patients should become MRD negative early after transplant,23,24 though one study found no association between early MRD-positivity at day +28 post-allo-HCT and survival.25 In most studies, persistent NPM1mut transcripts after transplant were associated with a statistically significant increased incidence of relapse and NPM1mut-based MRD monitoring preceded other MRD markers in impending relapse.23,24,26,27 Levels >0.1% NPM1mut/ABL1 beyond day +60 post-allo-HCT are translated into increased frequency of relapses, whereas the most powerful independent prognostic cutoff level for reduced survival was 10% NPM1mut/ABL1.25,28 MRD persistence at 3 months post-allo-HCT evaluated by deep targeted sequencing (variant allele frequency [VAF] >0.02%) was significantly and independently associated with increased relapse rates, suggesting that conversion to MRD-negativity by day 100 after allo-HCT is a precondition for achievement of stable remission.23 Since NPM1mut relapses tend to occur late (>6 months) after allo-HCT, our current everyday clinical practice is close MRD monitoring by mutant-specific RT-qPCR (sensitivity 10−4 to 10−5) in BM every 3 months.3 Preemptive interventions are considered for patients with persistent MRD >0.1% in 3 consecutive measurements or MRD >1% when confirmed in a repeated analysis within 4 weeks. MRD >10% (1000 NPM1mut/104ABL1 copies) is an indication for an intermediate intervention, since these patient relapse within a short period of time.28
Case 2
A 19-year-old male with a previously diagnosed t(8;21) core-binding factor (CBF) AML relapsed with 5567 RUNX1-RUNX1T1/104ABL1 copies in BM and underwent an HLA-matched sibling MAC-PBSCT in CR 2. BM at 3 months revealed 78 RUNX1-RUNX1T1/104ABL1 transcripts, while FC did not detect any leukemic cells. Cyclosporine was discontinued at day +145. BM at day +190 showed MRD persistence at a low level (4.3 RUNX1-RUNX1T1/104ABL1 copies). Chimerism assays of BM and PB indicated CC.
CBF fusion transcript monitoring
Though it is expected that CBF-positive nonleukemia cells should be cleared in the context of allo-HCT, CBF-fusion transcripts at low levels have been found to persist in long-term transplant survivors.29 A >3-log reduction in RUNX1-RUNX1T1 transcripts at any time point and >4-log reduction at 12 months post-allo-HCT are considered safe cutoff-levels for continuous CR. Increase of RUNX1-RUNX1T1 transcripts >1-log predicted relapse despite DLI, whereas patients with ≤1-log increase could be rescued with DLI.30,31 Similar MRD kinetics after transplant have been found in patients with CBFB-MYH11 AML.32,33 We quantify CBF-fusion transcripts by RT-qPCR in BM at 3 months posttransplant and adjust further MRD-monitoring intervals according to the MRD level detected. Preemptive interventions are considered for patients with persistent MRD >1% RUNX1–RUNX1T1 or CBFB-MYH11/ABL1 in 2 consecutive measurements or with >0.5-log increase of the CBF fusions in repeated analyses.
Case 3
A 21-year-old female patient with a previous history of Ewing sarcoma at age of 17 developed a therapy-related t(11;19)(q23;p13.1) AML. She underwent a MAC-BM transplantation from her 4/8 HLA-matched haploidentical mother with posttransplant cyclophosphamide (PTCY). KMT2A (previously termed mixed lineage leukemia [MLL])-ELL fusion transcripts in BM pretransplant evaluated by nested PCR (sensitivity 10−4) were undetectable. BM-MRD for MLL-ELL transcripts at 3 months was negative but was found positive at routine analysis on day +203 and in a repeated analysis 3 weeks later. Donor chimerism fell to 90% in the MRD-positive BM samples, whereas FC did not detect residual leukemic blasts.
MLL monitoring
Liu et al found that any MLL expression (>0.0%) after allo-HCT was highly predictive for relapse in multivariate analysis (>90% patients relapsed, hazard ratio [HR], 18.643).34 Most MRD positivity emerged at 3 to 5 months posttransplant, and the median time to relapse after MLL detection was 109 days. In contrast, <10% of the patients with undetectable MLL fusion transcripts relapsed. We routinely screen BM every 3 months for patient-specific MLL rearrangements by nested PCR (sensitivity 10−4), and any MRD positivity confirmed by repeated analysis is considered as a trigger for a preemptive intervention.
Case 4
A 37-year-old man with a FLT3-ITDmut AML achieved CR after 2 cycles of induction chemotherapy and received 2 additional consolidation cycles combined with midostaurin. He underwent an HLA-matched sibling allogeneic MAC-PBSCT with cyclosporine and methotrexate. At that time, the randomized trial of gilteritinib maintenance was not yet recruiting at our institution (NCT02997202) and the patient’s medical insurance rejected coverage for “off-label” sorafenib prophylaxis. Routine chimerism studies by STR-based PCR at day +37 depicted CC in BM, PB, and circulating T cells (> 95%). However, PB donor chimerism progressively decreased to 70% (day +130), despite fast weaning of cyclosporine from day +70 onwards. BM sampling on day +95 and day +130 showed 80% and 70% donor chimerism in the mononuclear fractions and 70% in the magnetic-bead–isolated CD34+ cell subset. BM-FC was negative for leukemic blasts.
FLT3-ITDmut monitoring
Case 5
A 64-year-old male with a MDS with excess of blasts (18%), complex karyotype, and mutated DNMT3 gene received 4 cycles of 5-azacytidine and underwent a reduced-toxicity conditioning (fludarabine, carmustine, and melphalan) PBSCT from an 23-year-old unrelated 10/10 HLA-matched donor.37 At transplant, 4-color FC showed persistence of 4% CD34+/CD117+/HLA-DR+ blasts with aberrant expression of CD7+. Routine BM evaluation at day+32 and day+105 showed CC (>95%). PB T-cell chimerism was constantly mixed (80% to 90% donor). Cyclosporine was discontinued at day +158. Progressive thrombocytopenia occurred after day +230, in parallel with new-onset PB mixed chimerism (70% donor). The loss of donor chimerism was confirmed in subsequent analyses in BM and PB mononuclear and polymorphonuclear fractions. FC in BM detected a 1.1% CD34+/CD117+ but CD7− blast population.
Chimerism monitoring
Chimerism analysis detects host-derived genetic material that cannot be equated with persistence of leukemia cells. Low-level host DNA (<1%) can be found for a longer period after transplant (eg, due to recipient stromal cells),38 bringing into question the clinical utility of the highly sensitive variant-allele–specific RT-qPCR–based or droplet digital PCR (ddPCR) techniques (sensitivity 0.1% to 0.01%) as compared with the standard STR-based PCR (sensitivity 1% to 5% according to microsatellite marker).39-42 Fluctuating low-level mixed chimerism in PB at the early posttransplant phase may be due to viral infections and expansion of residual, recipient-derived, virus-specific T cells.41 Chimerism patterns, kinetics, and clinical interpretation are strongly related to the transplant practice (eg, MAC vs reduced-intensity conditioning [RIC], T-cell depletion vs none) and to the underlying disease (eg, malignant vs nonmalignant).43-45 Lineage-specific chimerism analysis may increase its specificity in predicting relapse.46,47 Mixed chimerism in T cells is common after T-cell–depleted or RIC transplants and is weakly associated with later disease relapse.45 A prospective study showed that the decrease of CD34+-specific donor chimerism to <80% had 100% sensitivity and 86% accuracy in predicting relapse. This contrasts with the 14% sensitivity and 46% accuracy of conventional chimerism testing.48 As a general rule, the speed and extent of the decrease of donor chimerism predicts relapse with a higher specificity than a static approach considering chimerism levels only at individual time points.41 We routinely monitor chimerism by STR-PCR in PB every 2 weeks within the first 3 months and at least monthly thereafter. We additionally check monthly chimerism in ficoll-isolated mononuclear and polymorphonuclear fractions and in T cells isolated by magnetic beads.39 BM-chimerism analysis is routinely performed on days +30, +100, +270, and +365. A continuous decrease of donor chimerism in the non–T-cell compartment or drop of donor chimerism to <80% in the magnetic-bead–isolated CD34+ subsets raises suspicion for impending relapse. Our decision making for a preemptive intervention encompasses other MRD markers and clinical parameters (eg, pretransplant risk assessment, new-onset cytopenia).
FC monitoring
FC is hampered by its low sensitivity (10−2 to 10−4) and the many factors that can cause false-negative results, such as number of events analyzed, sample processing, hemodilution, and immunophenotypic switch.4 Data analysis and interpretation requires considerable expertise and experience to differentiate “leukemia-associated immunophenotype” or “different-from-normal” populations from regenerating BM cells.49 In expert hands, FC-MRD can be very specific, as demonstrated by Jacobsohn et al, who sorted the FC-detected suspicious cells and proved that these were of recipient origin and contributed to later morphological relapse.50 By applying a highly sensitive (0.1%) 10-color/1-million-events FC-MRD protocol, Zhou et al found that early (day +30) FC-MRD evaluation after allo-HCT has a very low ability to capture leukemic clones that emerge at later time points.51 The existing studies indicate that a new-onset FC-MRD positivity identifies patients with a high relapse risk.52-56 However, there is considerable variation in the protocols used, which makes it difficult to compare results among different studies or implement the results in one’s own laboratory. Most routine laboratories, including ours, will be able to measure FC-MRD at a level of 1%. We categorize FC-MRD results as “suspicious” or “positive” depending on the detected abnormal blast population.
WT1 monitoring
WT1 is a nearly universal leukemia antigen that can be measured in PB but is also overexpressed in normal regenerating BM cells. Patients who do not clear their pretransplant high BM-WT1 transcripts (>250 copies) at 3 months post-allo-HCT or who show a continuous increase of PB-WT1 transcripts are at risk for relapse.57,58 Patients with sustained low WT1 levels after allo-HCT (BM <100, PB <50 copies) had excellent outcomes.58-60 We have recently commenced PB-WT1 testing using a commercially available European Leukemia Net–certified RT-qPCR kit as part of developing release criteria for administration of WT1-specific T cells within a forthcoming multicenter study.61,62
DNMT3mut monitoring
The investigation of DTA mutations (ie, mutations in DNMT3, TET2, and ASXL1) after transplant may confront us with the engraftment of donor clonal hematopoiesis of indeterminate potential (CHIP).63 DNMT3-mutated host-derived CHIP is typically cleared (VAF <0.2%) at 3 months post-allo-HCT.64 Nakamura et al followed DNMT3 mutations (and other putative founder mutations) using a very sensitive (0.04%) ddPCR and found that increasing MRD positivity between 1 and 3 months post-allo-HCT was the most precise independent predictor of relapse (HR, 28.47; P < .0001).65 Of note, the DNMT3 mutations were found in the relapsed samples. Thus, in contrast to the nontransplant setting, where the persistence of DNMT3 mutations indicates remaining CHIP with no prognostic value,66 the above-mentioned study associates “nondonor” DNMT3 mutations posttransplant with the persistence of transformed DNMT3mut leukemic clones and increased relapse risk.65 The results from a similar prospective study are awaited to clarify this observation (NCT02872662). Another interesting finding of the Nakamura et al study is that the predictive utility of “noninvasive” MRD monitoring using serum DNA was comparable to that of MRD testing in BM.65
NGS-based monitoring
A persistent VAF >0.2% mutational burden in BM at day +21 is associated with an increased relapse risk (HR, 4.75).64 However, in nearly half of the patients, the day +21–detected mutations were stepwise cleared by 6 months (potentially mediated by GVL), and these patients did not relapse. Thus, the positive predictive ability for relapse of early NGS-MRD remains poor and is also hampered by the high sequencing error rate. Interestingly, mutations detected by NGS at day +21 expanded at relapse in relapsed patients.64
Case 6
A 34-year-old man with relapsed Philadelphia-chromosome–negative CD19+/CD10+ B- cell ALL was referred 16 months after initial diagnosis for a 9/10 HLA-matched unrelated MAC-PBSCT (12-Gy total body irradiation/etoposide) in CR 2. MRD monitoring was performed with Ig/TCR semi-quantitative PCR (sensitivity 10−4). At transplant, MRD was positive (10−3 to 10−4). FC in BM at day +41 revealed CD19+/CD10+/CD45dim low-side-scatter cells, well matched with regenerating precursors (hematogones). Ig/TCR-MRD in BM at 3 months was positive (10−3 to 10−4) and confirmed to be positive at the same level 2 and 4 weeks later. BM, PB, and T-cell fractions showed CC.
ALL monitoring
FC-based MRD monitoring in ALL post-allo-HCT is even more troublesome than in AML, since regenerating hematogones and ALL blasts share the same immunophenotype (CD19+/CD10+).67 Furthermore, amplification of comparable sequences in regenerated B/T cells may cause false-positive Ig/TCR results.68 IgH-V(D)J-NGS has better specificity and may also capture apparent clonal evolution.69,70 Early (day +30) Ig/TCR MRD positivity has low accuracy to predict a future relapse.71-73 At later time points, MRD levels >10−4 are associated with increased relapse rates but can be cleared by immunotherapy.71-74 In one series, patients with Ig/TCR MRD >10−3 or those who experienced >1-log MRD increase ultimately relapsed, regardless of immunosuppression discontinuation or DLI.72 Our clinical practice dictates that ALL patients should at least every other month monitored with Ig/TCR PCR (or NGS if feasible). Any MRD positivity beyond day +100 is a trigger for intervention. In case of >1-log increase of MRD or MRD level >10−2, we consider “off-label” blinatumomab or inotuzumab in line with phase 2 ongoing studies (eg, NCT03109093 and NCT04044560).
MRD-directed interventions after transplantation
Withdrawal of immune suppression
The burden of immunosuppression administered peri- and posttransplant is associated with risk of relapse.75,76 Withdrawal of immunosuppression is routinely considered as the first therapeutic maneuver for patients with posttransplant MRD status and without GVHD. Different from chronic myeloid leukemia, where impressive responses have been seen, immunosuppression withdrawal by itself is unlikely to result in a clinically significant benefit.46,77
DLIs
After withdrawal of immunosuppression, infusion of donor cells represents the standard approach for boosting the GVL effect.78 The efficacy of DLI has been demonstrated by their ability to prevent relapse or to treat overt relapse in some cases.79-82 In NPM1mut AML, DLIs have been shown to generate T cells with specificity against the mutant NPM1 protein.83,84 Published data on preemptive DLI for MRD positivity are limited to retrospective or nonrandomized prospective series in which the DLI-treated patients most likely represent a positively selected cohort (Table 2). MRD-WT1–triggered preemptive DLI could prevent relapse, especially when these were given at low (<100), but not higher (180), WT1 copies.85,86 The largest series of preemptive DLI was reported by a Chinese group, who prospectively monitored 814 patients (AML 70%) using FC- and WT1-based MRD. Administration of modified DLIs (see below) reduced 2.3-fold the relapse risk as compared with MRD-positive patients with no DLI.87 A preventive effect of DLI on relapse has also been suggested when the MRD trigger was chimerism loss or increased RUNX1-RUNX1T1 transcript levels.30,43
The optimal administration schedule for DLI in the preemptive setting is uncertain. An important safety issue that emerged from studies in which DLIs were given prophylactically (mostly in T-cell–depleted allo-HCT) is that the patient should be free of GVHD for at least 1 month and that the starting dose should be 1-log lower (0.5-1 × 106 CD3+ cells/kg) than the one used in the relapse setting.88 Most groups will give repetitive, dose-escalated (by a 2 to 5-fold increment) unmanipulated donor cells in 4- to 12-week intervals, but data are insufficient for clear recommendation. Future research is needed to determine the dose intensity and total number infusions that are necessary to achieve long-term remission. Similarly, further work is necessary to confirm whether MRD negativity is a sufficient end point or whether additional therapy, potentially including a second transplantation, is needed to secure a cure. As a practical measure, we routinely freeze and use donor cells isolated from the initial granulocyte colony-stimulating factor–mobilized graft, which have been shown to possess similar activity as unstimulated DLI.89 We reevaluate MRD after 2 to 4 cycles of DLI, and we “consolidate” by MRD response with an additional 4 to 8 infusions, provided that the patient is free of GVHD.
Haplo-DLI
Haplo-DLI may pose an increased risk for GVHD due to the high donor-recipient HLA disparity. In T-cell–depleted/CD34+-selected haploidentical allo-HCT, the CD3+ threshold for GVHD has been set at 3 × 104 cells/kg.90 Zalmoxis and ATIR101 are suicide-gene–engineered and alloreactive T-cell–depleted haplo-DLI products, respectively given prophylactically to enhance immune reconstitution.91,92 For preemptive interventions after anti-thymocyte globulin (ATG)-based haploidentical allo-HCT, the Chinese groups use granulocyte colony-stimulating factor–mobilized haplo-DLI followed by short-term immunosuppression (termed modified-DLIs).20 After PTCY-based haploidentical allo-HCT, prophylactically or preemptively administered unmanipulated haplo-DLI with a starting dose of 1 × 104 to 1 × 105 CD3+ cells/kg (ie, 1-2 log lower than from HLA-matched donor) were relative safe.93-96 There are 25 cases of MRD-triggered haplo-DLI after PTCY haploidentical allo-HCT reported so far, of which nearly 50% responded (normalized WT1, negativity of leukemia-specific marker, increase of donor chimerism).93,94 With the increasing use of haploidentical transplants, it must be noticed that nearly one-third of relapses that occur after mismatched related transplants (and nearly 10% of relapses after unrelated allo-HCT) show genomic loss of the mismatched HLA haplotype in which the use of DLIs will probably have no effect.12,97 Thus, documentation of “HLA-loss” at posttransplant relapses has relevant therapeutic implications and can be performed in nonpurified samples by the use of the HLA-KMR qPCR (GenDx, Utrecht, The Netherlands).98
Pharmacologic intervention with or without DLI after transplantation
Pharmacological intervention for posttransplant MRD aims to mediate a direct antileukemic effect and/or to enhance the alloreactive response. There is convincing evidence that immunomodulators alone cannot facilitate long-term survival, but this can be achieved when they are combined with DLI.81 HMAs (azacitidine and decitabine) may beneficially influence the balance between GVL and GVHD by enhancing the immunological visibility of leukemia cells (eg, through expression of silenced cancer/testis antigens and activation of interferon responses) while mitigating GVHD through expansion of regulatory T cells.99-101 However, it is also possible that HMAs may hamper GVL responses by inhibiting the function of natural killer cells or increasing the frequency of regulatory T cells in the BM.102,103 Two prospective studies in AML and MDS demonstrated that azacitidine post-allo-HCT could safely and effectively treat MRD (conversion of loss of CD34+ donor chimerism, NPM1mut or RUNX1-RUNX1T1 positivity), ultimately leading to prevention of relapse in approximately one-third of patients.104,105 Better results were found when azacitidine was given together with DLIs.106 Currently, a prospective randomized controlled study compares the safety and efficacy of MRD-triggered HMA+DLI versus DLI alone (NCT03662087). Our preemptive protocol for MRD positivity in AML and MDS entails 4 cycles of azacitidine (32-75 mg/m2, days 1-5) followed by a fixed dose of DLI at day +14. MRD responders are scheduled to receive 4 more cycles, and nonresponders receive escalated doses of DLI after each azacitidine cycle.
Interferon-α and interleukin-2 alone or together with DLIs have also been tested as immunomodulators in the MRD preemptive setting, but with doubtful effects and safety concerns.87,107 Lenalidomide given as maintenance therapy after allo-HCT has been associated with severe GVHD, but it can be more safely administered when combined with azacytidine.108,109 Extended azacitidine dosing using an oral formulation of the drug (CC-486) and panobinostat (deacetylase inhibitor) have shown promising results in prophylactic phase 1/2 studies.110,111 Case series reported the efficacy of immune checkpoint inhibitors (anti-CTLA-4 and anti-PD-1/PD-L1) in relapsed disease, but their use is associated with high rates of severe, often life-threatening GVHD and thus the administration of these drugs in the preemptive MRD setting is not justified outside a clinical trial.112,113
Given the positive results in many retrospective and prospective studies, most FLT3-ITDmut AML patients today will receive maintenance therapy with an FLT3 inhibitor, either off-label or within a clinical trial. Given the possible synergism between FLT3 inhibitors and alloreactive donor T cells, we favor the combination of sorafenib with DLI in patients with FLT3-ITDmut AML needing preemptive therapy.114 The isocitrate dehydrogenase inhibitors (ivosidenib and enasidenib) are currently tested in isocitrate dehydrogenase–mutated AML as maintenance and salvage therapy after allo-HCT.
How did we intervene?
No preemptive intervention was undertaken for patient 2. Further BM aspirations were scheduled on days +270 and +365 after allo-HCT, which revealed 0.08% and 0.00% RUNX1-RUNX1T1/104ABL1 copies, respectively. We administrated DLI in the remaining patients. The starting DLI dose was 1 × 106 CD3+ cells/kg for patients 1, 5, and 6; 0.5 × 106 CD3+ cells/kg (<4 weeks after discontinuation of immunosuppression) for patient 4; and 1 × 104 CD3+ cells/kg (haploidentical donor) for patient 3. We combined DLI with sorafenib in case 4 (FLT3-ITDmut) and with azacitidine in case 5 (MDS). In patient 1 (NPM1mut AML), NPM1mut transcripts were no longer detectable in the BM after 3 DLI cycles. We gave in total 6 escalated DLIs (maximal dose, 5 × 106 CD3+ cells/kg) without complications, and the patient is now 4.5 years after transplantation and in good health. Patient 3 (MLL-ELL+ AML) relapsed after the third haplo-DLI (1 × 105 CD3+ cells/kg) and succumbed to her disease 371 days after transplant. Patient 4 (FLT3-ITDmut AML) received an initial total dose of 800 mg/day sorafenib, but this was reduced to 400 mg/day due to high serum amylase levels. After 3 months of sorafenib combined with DLI, the PB chimerism converted to completely donor. He received in total 6 DLIs and continues on sorafenib at 1.5 years after allo-HCT. Patient 5 (MDS) received in total 8 cycles of azacitidine (75 mg/m2 on days 1-5) followed by a fixed dose of DLI (1 × 106 CD3+ cells/ kg). The PB and BM mixed chimerism switched to CC after the fourth azacitidine/DLI cycle. He is now 17 months after the start of preemptive therapy with a good performance status. In patient 6 (ALL), the BM-MRD assessment was negative after the fourth DLI given at 5 × 106 CD3+ cells/kg. After the fifth DLI (1 × 107 CD3+ cells/kg), the patient developed a biopsy-proven liver GVHD (hepatitic variant), which was resolved after treatment with steroids and budesonide. The patient is now 3.2 years after transplantation with a good performance status. Preemptive MRD-triggered intervention, although not effective, was probably a correct decision in case 3. In cases 1, 4, 5, and 6, we cannot be sure whether we “overreacted.”
Summary
Dynamic MRD monitoring after allo-HCT may improve outcomes, but current existing data do not facilitate a clear recommendation for a standardized pathway for MRD testing and MRD-directed intervention after transplant. A significant challenge will be to perform well-designed prospective clinical trials in these relatively small patient populations. Although most relapses occur within the first year after transplantation, posttransplant surveillance should probably be continued for up to 2 years or beyond.3 This is especially true for haploidentical transplants in which the frequently observed “HLA-loss” relapses tend to occur at later time points.97 Technical aspects of MRD monitoring according to sample and target analyzed have been provided by the European Leukemia Net.4 BM-MRD is in general 1-log more sensitive than PB and should be routinely performed; however, in the context of allo-HCT, PB-chimerism studies can be used to adjust and better interpret other MRD test results (Figure 1).115 The MRD techniques continue to advance (eg, error-corrected NGS, sensitive ddPCR, and MRD from circulating DNA) and are expected to improve the accuracy of assessment of clonal and immunological changes in low-volume residual disease, thus enabling a more rational therapeutic intervention than is currently possible. In parallel, the landscape of cellular and targeted immunotherapy is evolving rapidly (eg, monoclonal antibodies, bispecific T-cell engagers, checkpoint inhibitors, antigen-specific T cells, chimeric antigen receptor and other genetically engineered T cells, and natural killer cells).116 Less progress has been made in monitoring the speed and quality of GVL reconstitution. Recent reports suggest that the increased frequency of regulatory T cells and exhausted leukemia-specific T cells in BM or the coexpression of inhibitory molecules on circulating T cells represents a dysfunctional GVL pattern that permits eventual relapse.103,117 Understanding the interplay between GVL and MRD remains a major challenge.
Acknowledgments
The author thanks Maria Gilleece and Panagiotis Tsirigotis for critical proofreading and editing of the manuscript and Maria Liga and Nikos Spyridis for their everyday collegial support.
Authorship
Contribution: A.S. performed literature research and wrote the manuscript.
Conflict-of-interest disclosure: A.S. declares no competing financial interests.
Correspondence: Alexandros Spyridonidis, Department of Internal Medicine, BMT Unit, University Hospital of Patras, 26500 Rio, Patras, Greece; e-mail: spyridonidis@upatras.gr.