Abstract
Steroid-resistant or steroid-refractory acute graft-versus-host disease (SR-aGVHD) poses one of the most vexing challenges faced by providers who care for patients after allogeneic hematopoietic cell transplantation. For the past 4 decades, research in the field has been driven by the premise that persistent graft-versus-host disease (GVHD) results from inadequate immunosuppression. Accordingly, most efforts to solve this problem have relied on retrospective or prospective studies testing agents that have direct or indirect immunosuppressive effects. Retrospective studies far outnumber prospective studies, and no controlled prospective trial has shown superior results for any agent over others. Truth be told, I do not know how to treat SR-aGVHD. Preclinical work during the past decade has provided fresh insights into the pathogenesis of acute GVHD, and translation of these insights toward development of more effective treatments for patients with SR-aGVHD has at last begun. Given the limited state of current knowledge, this “How I Treat” review highlights the overriding imperative to avoid harm in caring for patients with SR-aGVHD. Prospective trials that are widely available are urgently needed to advance the field.
Introduction
Despite prophylactic immunosuppression after allogeneic hematopoietic cell transplantation (HCT), ∼30% to 70% of patients require additional systemic treatment of acute graft-versus-host disease (GVHD), most often with prednisone at 1.0 to 2.0 mg/kg per day.1 In approximately one third of cases, the disease shows no improvement after 4 weeks of treatment.2 To date, no consensus has been reached regarding the optimal approach for management of steroid-resistant or steroid-refractory acute graft-versus-host disease (SR-aGVHD).3 The choice of treatment has been guided largely through empirical trial and error according to physician experience, ease of use, need for monitoring, risk of toxicity, and potential exacerbation of preexisting comorbidity.
This review outlines the general principles that guide my management of patients with SR-aGVHD, discusses the potential application of blood biomarkers to improve current management, summarizes evidence suggesting the treatment agents that might be preferred over others, and highlights mechanisms that could explain why acute GVHD does not always respond to initial treatment with glucocorticoids.
Case summary
A 44-year-old man received a mobilized blood cell graft from an HLA-identical sibling after reduced intensity conditioning for treatment of acute myeloid leukemia in first remission. He received tacrolimus and mycophenolate mofetil for immunosuppression after HCT. His recovery was complicated by persistent anorexia, nausea and vomiting without diarrhea, rash, or abnormal liver function tests, prompting empiric treatment with prednisone at 0.5 mg/kg per day and oral beclomethasone on day 32 for presumed upper gastrointestinal GVHD. He subsequently developed diarrhea and, on day 39, imaging showed mild wall thickening of the terminal ileum. On day 41, the dose of prednisone was increased to 2.0 mg/kg per day. On day 45, the dose of prednisone was decreased to 1.0 mg/kg per day after laboratory tests showed toxigenic Clostridium difficile infection, which was treated with oral vancomycin and metronidazole. Follow-up imaging on day 46 showed an increase in the length of bowel wall thickening and mucosal hyperenhancement in the terminal half of the ileum, and endoscopic biopsies on day 47 showed mild histologic GVHD activity in the duodenum and minimal histologic GVHD activity in the sigmoid colon and rectum. Therefore, the dose of prednisone was increased to 2.0 mg/kg per day on day 48.
Large-volume diarrhea persisted with new abdominal pain, prompting enrollment in a clinical trial testing the efficacy and tolerability of α-1 antitrypsin for treatment of SR-aGVHD. α-1 Antitrypsin was administered from days 50 through 64. Prednisone doses were tapered by 0.2 mg/kg every 3 days between days 56 and 61, but tapering was discontinued because follow-up endoscopy on day 61 showed new histologic GVHD activity in the gastric antrum and increased activity in the duodenum. Symptoms improved during treatment with a total of 6 mg/kg rabbit anti-thymocyte globulin (ATG) (Thymoglobulin; the only formulation marketed in the United States) between days 63 and 75. He was also treated with lithium carbonate from days 63 through 80 to promote mucosal regeneration, and diarrhea was managed symptomatically with octreotide and tincture of opium. At the onset of treatment with ATG, the prednisone dose was decreased to 1.5 mg/kg per day on day 63 and to 1.0 mg/kg per day on day 65 to decrease the risk of opportunistic infection. He was also treated with mold-active antifungal agents, acyclovir, and dapsone to decrease the risk of opportunistic infection. When he returned to the care of his referring physician on day 104, he had no evidence of chronic GVHD. The team recommended weekly screening for cytomegalovirus, Epstein-Barr virus, and adenovirus activation for ≥6 months after treatment with ATG and until the absolute lymphocyte count surpassed 300 per microliter.
Treatment of acute GVHD continued with tacrolimus, oral beclomethasone and budesonide, and prednisone at gradually tapering doses. At 17 months after HCT, the prednisone dose reached 7.5 mg/d. After replacement of prednisone with hydrocortisone, he developed nausea, vomiting, diarrhea with bright red blood, and abdominal pain. Treatment was resumed with prednisone doses increased step-wise to 2 mg/kg per day without improvement. Endoscopic biopsy of the colon on day 509 showed crypt dropout representing >50% of the area sampled, with frequent apoptotic bodies in remaining crypts, confirming the diagnosis of recurrent acute GVHD.
Upon his readmission to our hospital on day 525, sigmoidoscopy showed extensively denuded mucosa throughout the visualized colon, and histopathology showed necrotizing enterocolitis, denuded colonic mucosa with focal regeneration, and occasional apoptotic epithelial cells consistent with GVHD (Figure 1). I considered the use of ATG for management of SR-aGVHD, but because adenovirus activation was detected in the blood, I substituted sirolimus for tacrolimus, continued treatment with oral beclomethasone and budesonide and high-dose prednisone, and added lithium carbonate at doses adjusted to maintain trough serum concentrations at 0.5 to 0.8 mEq/L. Adenovirus infection was treated with weekly cidofovir, ending on day 558.
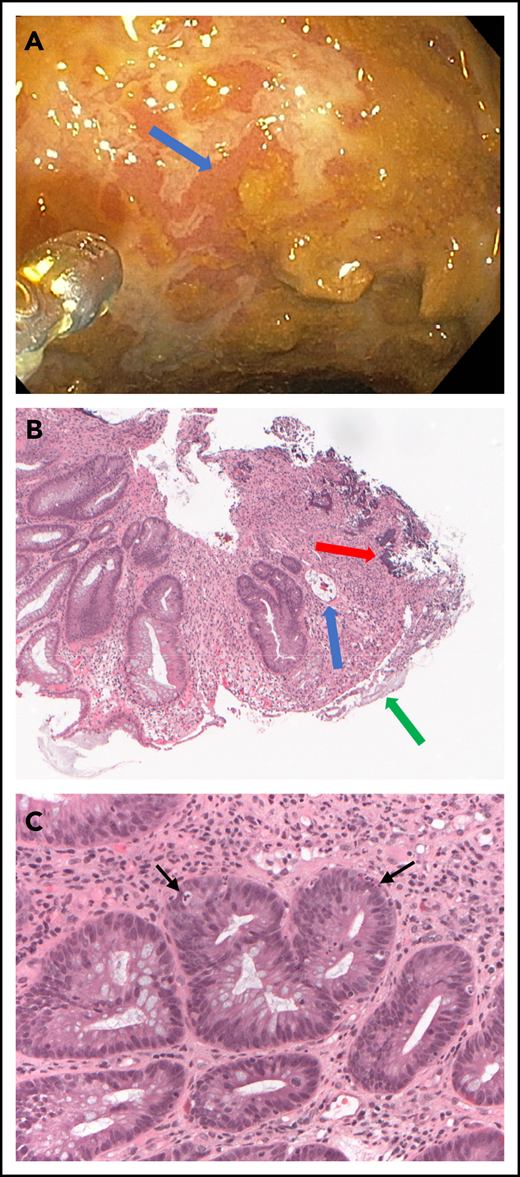
Endoscopic and pathologic evaluation of the sigmoid colon on day 525 after transplantation. (A) Endoscopic view of the colon shows serpiginous ulceration of the mucosal surface (arrow). (B) Low-power view of colon biopsy shows atypical glands indicating regeneration, intramural bacteria (red arrow), fibrinous debris coating denuded luminal surface (green arrow), and an exploded crypt lacking viable cells (blue arrow). Hematoxylin and eosin, original magnification, ×5. (C) High-power view shows crypts with apoptotic cells (arrows) and intense inflammatory infiltrate surrounding the crypts. Hematoxylin and eosin, original magnification, ×20.
Endoscopic and pathologic evaluation of the sigmoid colon on day 525 after transplantation. (A) Endoscopic view of the colon shows serpiginous ulceration of the mucosal surface (arrow). (B) Low-power view of colon biopsy shows atypical glands indicating regeneration, intramural bacteria (red arrow), fibrinous debris coating denuded luminal surface (green arrow), and an exploded crypt lacking viable cells (blue arrow). Hematoxylin and eosin, original magnification, ×5. (C) High-power view shows crypts with apoptotic cells (arrows) and intense inflammatory infiltrate surrounding the crypts. Hematoxylin and eosin, original magnification, ×20.
His hospital course was complicated by C difficile infection diagnosed on day 527, which was treated with oral vancomycin. On day 535, he presented with colonic perforation requiring resection of the proximal sigmoid colon with a diverting colostomy. He was treated with IV vancomycin, and prednisone doses were tapered beginning on day 536 to facilitate wound healing after surgery. He was discharged to the care of his referring physician on day 580 after 3 weeks of inpatient physical rehabilitation. At hospital discharge, the prednisone dose was 30 mg and 25 mg on alternating days. Doses of prednisone were tapered as tolerated by abdominal symptoms. Treatment with sirolimus was complicated by low-grade thrombotic microangiopathy; on day 573, tacrolimus was substituted for sirolimus. He has survived beyond 4.2 years after HCT, continuing treatment with prednisone at 8 mg/d, tacrolimus, and budesonide, with no evidence of acute or chronic GVHD.
Principles illustrated by the case
This case is atypical of SR-aGVHD in its severity and complications, its recurrence >1 year after HCT, and, most strikingly, in the patient’s survival. Most cases occur within the first 100 days, mucosal denudation occurs infrequently, and most patients with intestinal mucosal denudation due to acute GVHD die within 6 months after the diagnosis.4 On the other hand, this case is typical in its involvement of the gastrointestinal tract, although SR-aGVHD can involve the skin and liver, with or without gastrointestinal involvement. The case illustrates many of the general principles that guide my approach to treatment of SR-aGVHD.
I use the lowest effective dose of prednisone or methylprednisolone
High-dose glucocorticoid treatment has many well-recognized toxic effects, including hyperglycemia, hypertension, fluid retention, and sleep and mood disturbances. Proximal myopathy is one of the more insidious and disabling toxicities of long-term treatment, and it is often exacerbated by lack of physical activity. I use physical therapy as a measure to offset myopathy.
I use topical therapy to supplement systemic treatment
For example, psoralen and UV-A exposure, topical corticosteroid creams, and topical tacrolimus can help to control skin involvement. I use oral budesonide together with beclomethasone compounded in our pharmacy for treatment of gastrointestinal involvement in nearly all patients. I acknowledge, however, that a randomized double-blind trial to evaluate this practice as an adjunct to initial treatment did not meet its primary end point,5 and this practice has never been formally evaluated in patients with steroid-refractory acute gastrointestinal GVHD. These agents have potent local effect in the intestine with low systemic effect because of rapid metabolic inactivation in the liver.
I use follow-up endoscopy to evaluate the response to treatment in patients with persistent diarrhea
An effective treatment would be expected to produce histologic improvement before epithelial repair has improved intestinal function. Absence of clinical improvement or histologic improvement as demonstrated by endoscopic biopsy6 after 2 weeks would suggest that treatment should be changed.
I enroll patients in clinical trials whenever possible
Despite the many gaps in knowledge, however, few such trials are available, as discussed in the section titled "Selection of treatment of SR-aGVHD."
I am mindful of causes other than GVHD that could exacerbate symptoms
In patients with diarrhea, these include C difficile infection, viral infections (eg, cytomegalovirus, adenovirus, rotavirus, and norovirus), and bacterial infections.
I use prophylactic medications to prevent opportunistic infection
These include mold-active antifungal agents (eg, voriconazole or posaconazole), antiviral agents (eg, acyclovir or valacyclovir) to prevent herpes simplex and varicella zoster infection, trimethoprim-sulfa, dapsone, atovaquone, or pentamidine to prevent Pneumocystis infection, and antibiotics (eg, levofloxacin) to prevent bacterial infection when the absolute neutrophil count is <500 per microliter. In addition, I screen at weekly intervals for early detection of cytomegalovirus, adenovirus, or Epstein Barr virus activation when immune function is severely impaired, especially after treatment with high-dose rabbit ATG. Patients with severe hypogammaglobulinemia may benefit from replacement therapy.
I avoid excessive immunosuppression with multiple agents given concurrently or with high doses of rabbit ATG
Some retrospective studies have shown high response rates in patients treated with rabbit ATG, but this benefit can come at the cost of low survival because of opportunistic infections.7,8 In the current case, the dose of prednisone was decreased from 2.0 mg/kg per day to 1.0 mg/kg per day within 2 days after starting treatment with rabbit ATG, and the total dose of rabbit ATG was limited to 6 mg/kg. When acute GVHD recurred, I substituted sirolimus for tacrolimus instead of using both drugs together, and I avoided the use of rabbit ATG so that adenovirus infection could be controlled.
I use lithium to promote intestinal epithelial repair
This practice is supported by results of an unstructured prospective single-arm trial.4 Among other possible targets, lithium inhibits glycogen synthase kinase-3, a negative regulator of β-catenin.9 Inhibition of glycogen synthase kinase-3 by lithium promotes β-catenin–mediated transcription, which activates the Wnt pathway, thereby stimulating proliferation of intestinal epithelial stem cells. A retrospective time-dependent multivariable proportional hazards analysis showed that lithium treatment in patients with intestinal mucosal denudation as a complication of GVHD at our center was associated with a 72% decrease in the mortality hazard (hazard ratio, 0.28; 95% confidence interval, 0.15-0.54) (Gideon Steinbach, David Myerson, Ted A. Gooley, George B. McDonald, and Paul J. Martin, unpublished observations, 10 May 2019). In this study, all but 1 of the 30 patients not treated with lithium died within 6 months after the initial diagnosis of intestinal denudation. In some patients, treatment with lithium was discontinued because of fatigue, somnolence, confusion, or blunted affect.4 To minimize the risk of toxicity, I adjust doses of extended-release lithium carbonate to maintain trough serum concentrations between 0.5 and 0.8 mEq/L. The extent of benefit from lithium treatment in patients with or without intestinal mucosal denudation and with or without concurrent liver GVHD has not been evaluated in controlled prospective trials, and the use of lithium for this indication does not have regulatory approval.
I use other supportive care to ensure adequate nutrition and control of symptoms
Nutrition can be provided through parenteral, enteral, or oral feeding, and agents such as loperamide, octreotide, and tincture of opium may be helpful in controlling diarrhea.6,10
Potential use of biomarkers to improve the management of SR-aGVHD
The field has not reached consensus regarding the definition of SR-aGVHD. I would begin to recognize GVHD as steroid refractory when patients are treated with steroids at prednisone-equivalent doses ≥1 mg/kg day with worsening of GVHD manifestations across any interval ≥2 days before tapering of steroid doses has begun, have persistent grade 2-4 GVHD across any interval ≥7 days without improvement during continued steroid treatment at a prednisone-equivalent dose >0.4 mg/kg per day, or have initial improvement followed by exacerbation of GVHD across any interval ≥3 days during appropriately tapered glucocorticoid treatment at any prednisone-equivalent dose >0.4 mg/kg per day. I typically taper prednisone doses by 0.2 mg/kg per day every 5 days in patients with persistent manifestations and every 3 days in those without persistent manifestations. Decisions to begin second-line treatment depend on response and toxicity.11 Providers caring for patients who begin to develop myopathy should not hesitate to start treatment with second-line agents.
The MAGIC Consortium has advanced the field by identifying and validating the use of ST2 and REG3α as prognostic biomarkers predicting nonrelapse mortality (NRM) in patients with acute GVHD.12 They evaluated these biomarkers measured at day 7 after initial treatment in patients stratified according to the presence or absence of response and according to standard and high Minnesota risk score categories2 at day 7 (Table 1). The estimates of 0.29, 0.46, and 0.84 within each stratum of Table 1 represent the positive predictive values of the biomarker algorithm for predicting NRM at 1 year.
These observations add considerable refinement and nuance to the definition of SR-aGVHD. We previously showed that older patient age, overt gastrointestinal bleeding, and high serum total bilirubin concentrations predict a high risk for mortality in patients with SR-aGVHD.13 Interventions are unlikely to be effective in patients who have already developed such severe clinical manifestations. The biomarker studies provide a standard time point early after starting treatment when laboratory tests can be used to predict the risk of subsequent NRM. Most importantly, they provide predictions before GVHD-related damage has become irreversible, thereby affording an opportunity to intervene early with effective treatment.
The positive predictive values relate importantly to the tolerance for risk in testing new agents in patients with different baseline risks for NRM. Providers and patients would be much more willing to accept uncertainty and tolerate high anticipated risk with a novel clinical intervention if the 1-year probability of NRM were 0.84 than they would if the probability were only 0.29 (Figure 2). Conversely, a novel intervention with less uncertainty and much lower anticipated risk could be tested simultaneously in all patients with biomarker values above the threshold, although the opportunity to demonstrate measurably improved survival may be much greater in the high-risk stratum than in the lower-risk strata.
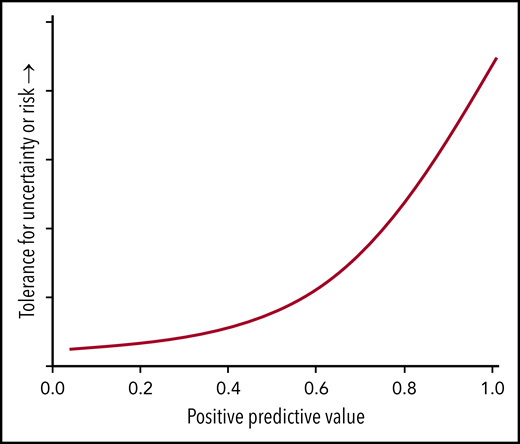
Higher positive predictive values justify clinical trials with greater uncertainty or higher anticipated risk. The positive predictive value indicates the proportion of patients with a positive test (eg, biomarker values above a preset threshold) who have a diagnosis or outcome predicted by the test (eg, death within 1 year). A high probability of death, as indicated by a positive test, makes it ethically acceptable to test interventions that have uncertain outcomes or higher anticipated risk. Such uncertainty or risk would not be ethically acceptable in situations with a low probability of death.
Higher positive predictive values justify clinical trials with greater uncertainty or higher anticipated risk. The positive predictive value indicates the proportion of patients with a positive test (eg, biomarker values above a preset threshold) who have a diagnosis or outcome predicted by the test (eg, death within 1 year). A high probability of death, as indicated by a positive test, makes it ethically acceptable to test interventions that have uncertain outcomes or higher anticipated risk. Such uncertainty or risk would not be ethically acceptable in situations with a low probability of death.
Selection of treatment of SR-aGVHD
The recent approval of ruxolitinib for treatment of SR-aGVHD in the United States has changed the landscape of interventions for this indication.14 The REACH-1 trial that led to this approval was a single-arm phase 2 study that enrolled 71 patients with a median age of 58 years.15 Sixty-eight percent of the patients had grade 3 or 4 GVHD at enrollment. At day 28, the complete response rate was 27%, and the overall response rate was 55%. NRM at 6 months was 44%, and overall survival at 6 months was 51%. Notable adverse events included anemia (65% of patients), thrombocytopenia (62%), neutropenia (48%), cytomegalovirus infection (13%), sepsis (13%), and bacteremia (10%).
Results of this study were comparable to aggregate outcomes summarized in the 2012 American Society of Blood and Marrow Transplantation (ASBMT) Guideline on treatment of acute GVHD.3 In that review of 29 curated retrospective and prospective studies of second-line or subsequent treatment of acute GVHD, the aggregate complete response rate was 32%, overall response rate was 58%, and survival at 6 months after treatment was 49%. Several key features of the REACH-1 study contributed to the regulatory approval. The eligibility criteria and definitions of SR-aGVHD ensured that continued steroid treatment was unlikely to control the disease. Ruxolitinib was the only systemic intervention allowed at enrollment. The primary end point of complete or partial response at day 28 was well defined, and patients who received additional systemic interventions for treatment of acute GVHD before day 28 were categorized as not having a response. With these design features, the treatment effect attributable to ruxolitinib was unambiguous. The toxicity of ruxolitinib had been well characterized in trials for other indications, and no unusual side effects were observed in patients with acute GVHD.
Although results of the REACH-1 trial showed that benefit from the treatment effect of ruxolitinib outweighed the harm from side effects, the data raise the question of whether outcomes with ruxolitinib are better than those with other agents that have been used to treat SR-aGVHD, a question to be answered by the REACH-2 trial (NCT02913261). To address this question, I reviewed retrospective and prospective studies that included ≥10 patients treated for SR-aGVHD published from 2012 to the present and not included in the ASBMT Guideline (Table 2). Because response is necessary, but not sufficient, for survival, and given the potential harms of treatment of SR-aGVHD, the analysis focuses exclusively on 6-month survival as the most important and unambiguous outcome measure. Unlike the 2012 ASBMT Guideline, reports were not evaluated for quality, and studies of investigational products were not excluded.
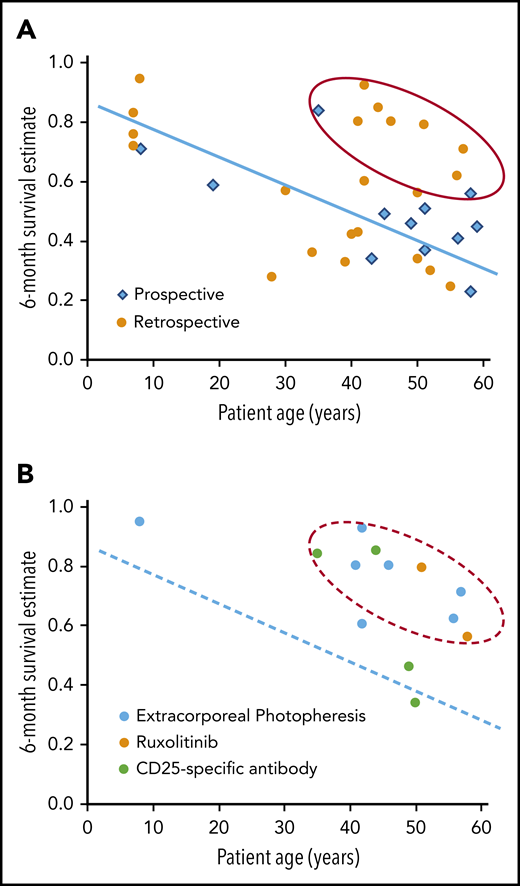
Figure 3 shows a correlation between median patient age and 6-month survival in studies that included ≥30 patients. This correlation was not apparent in the studies evaluated in the 2012 ASBMT Guideline, but a recent single-institution analysis showed a similar correlation between patient age and survival after second-line treatment of SR-aGVHD.16 Although the data have not been analyzed formally, the results in the figure suggest a group of 9 studies with better than expected 6-month survival compared with other studies with patient age in the same range. Agents tested in these studies included ruxolitinib (n = 2; 1 prospective),15,17 extracorporeal photopheresis (ECP) (n = 5; all retrospective),18-22 and CD25-specific antibodies (n = 2; 1 prospective).23,24
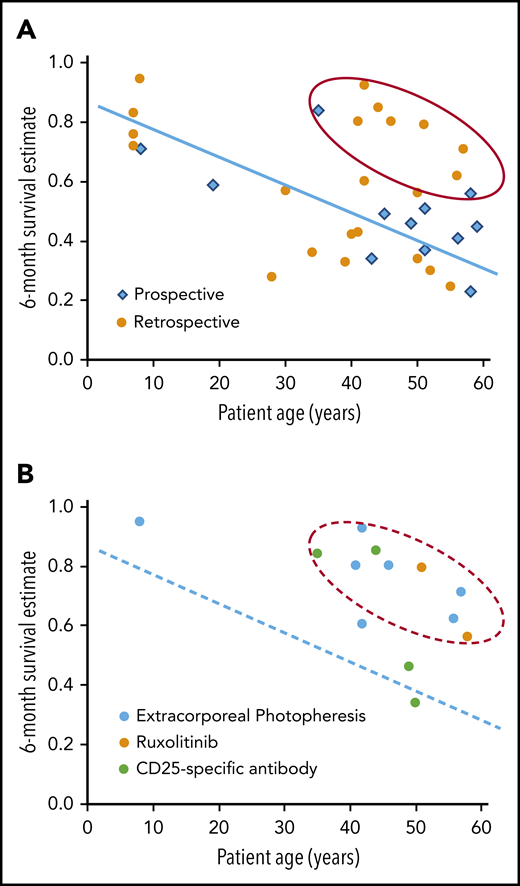
Older patient age is associated with lower survival at 6 months after treatment of SR-aGVHD. (A) The graph shows the correlation of median patient age with 6-month survival in retrospective and prospective studies of interventions to treat SR-aGVHD. Studies included ≥30 patients, were published from 2012 to the present, and were not included in the 2012 ASBMT Guideline (supplemental Table 1, available on the Blood Web site). The blue line and red oval are provided to help visualize the correlation and identify possible outliers with better than expected results, and they do not represent the results of a statistical analysis. (B) The graph highlights studies that tested ECP, ruxolitinib, or CD-25-specific antibody and included ≥30 patients. The dashed blue line and dashed red oval replicate the line and oval from Figure 3A.
Older patient age is associated with lower survival at 6 months after treatment of SR-aGVHD. (A) The graph shows the correlation of median patient age with 6-month survival in retrospective and prospective studies of interventions to treat SR-aGVHD. Studies included ≥30 patients, were published from 2012 to the present, and were not included in the 2012 ASBMT Guideline (supplemental Table 1, available on the Blood Web site). The blue line and red oval are provided to help visualize the correlation and identify possible outliers with better than expected results, and they do not represent the results of a statistical analysis. (B) The graph highlights studies that tested ECP, ruxolitinib, or CD-25-specific antibody and included ≥30 patients. The dashed blue line and dashed red oval replicate the line and oval from Figure 3A.
This informal analysis does not account for other factors that might influence survival in patients with SR-aGVHD, such as grade 3-4 severity at enrollment and the number of prior systemic treatments. Retrospective studies have many weaknesses and are better understood as descriptions of physician behavior more than any rigorous analysis of outcomes with a specific intervention. Even so, the patterns suggesting consistently favorable results with ruxolitinib and ECP would prompt me to use either of these agents preferentially in treating SR-aGVHD. For patients with active infection or severe neutropenia or thrombocytopenia, I would use ECP rather than ruxolitinib.25
Unfortunately, very few clinical trials are currently open for enrollment of patients with SR-aGVHD (Table 3). Agents studied in these trials include mesenchymal stem cells (n = 4), fecal microbiota transplantation (n = 2), ruxolitinib (n = 2, including 1 for children), neihulizumab (CD162-specific antibody, n = 1), and recombinant human gonadotropin (n = 1). Only 3 of these studies have industry sponsors, and 4 are open at only a single location. The only double-blind randomized trial of mesenchymal stem cells for treatment of SR-aGVHD showed no survival benefit when added to best available therapy.26 Whether this negative result is limited to this specific product or applies more broadly to other similar products remains to be determined.
The field would be advanced if the recent regulatory approval of ruxolitinib inspired academic and industry efforts to make clinical trials more widely available for patients with SR-aGVHD. One such trial could compare overall survival up to 1 year with the use of ruxolitinib vs ECP in adult patients with a predicted 1-year NRM risk of 0.84 or 0.46 based on response, Minnesota risk score, and ST2/REG3α biomarker values at day 7 after starting treatment with prednisone at doses ≥ 1 mg/kg per day.
Mechanisms that might contribute to SR-aGVHD
How donor T cells constantly stimulated by recipient alloantigens can persist and function without activation-induced apoptosis or anergy is unclear. Nonetheless, the approach to treatment of SR-aGVHD during the past 4 decades has been driven by the notion that the lack of response to initial treatment results from inadequate immunosuppression. Accordingly, most clinical trials have been designed to test agents that in 1 way or another intensify immunosuppression in the recipient.
A variety of mechanisms might contribute to the development of SR-aGVHD. Glucocorticoids induce Toll-like receptor 4–activated monocytes to express an intermediate phenotype that promotes development of proinflammatory T helper (Th)17 cells27,28 that, in turn, are resistant to glucocorticoid-induced apoptosis and suppression of cytokine expression.29,30 Patients with GVHD have high percentages of monocytes with this intermediate phenotype.27 Further evidence has suggested a transition from Th1 cell–based immune pathogenesis to Th17- and T cytotoxic (Tc)17-cell–based immune pathogenesis in the development of severe persistent SR-aGVHD.31,32 If so, identification of agents that interrupt induction of glucocorticoid resistance or have activity against glucocorticoid-resistant Th17 cells and Tc17 cells could advance the field.
Tissue biopsies and blood mononuclear cells from patients with severe GVHD have high numbers T cells that produce granulocyte-macrophage colony-stimulating factor (GM-CSF).33 In turn, GM-CSF drives GVHD pathology by licensing donor-derived phagocytes to produce inflammatory mediators, such as interleukin-1β (IL-1β) and reactive oxygen species. These results suggest that neutralization of GM-CSF might improve outcomes in patients with SR-aGVHD.
Solutions to the problem of SR-aGVHD might be found by elucidating mechanisms other than insufficient or ineffective immunosuppression.34 At least 4 hypotheses have drawn recent attention.
Impaired epithelial regeneration
The stem cell compartment is the primary intestinal target of allogeneic T cells during immune-mediated tissue damage after HCT.35 Our experience with the use of lithium to promote intestinal stem cell proliferation and epithelial regeneration in patients with denuded intestinal mucosa could encourage evaluation of other agents with similar effects. Preclinical studies have shown that treatment with the Wnt agonist R-spondin1 or with IL-22 in vivo enhanced the recovery of intestinal stem cells, increased epithelial regeneration, and reduced intestinal pathology and mortality in mice with GVHD.36,37 A phase 1/2 trial testing the safety, tolerability, and pharmacokinetics of IL-22 immunoglobulin G2 Fc in combination with systemic corticosteroids in patients with newly diagnosed acute lower gastrointestinal GVHD is in progress (NCT02406651).
Intestinal dysbiosis
Animal experiments have shown that gut microbiota contribute to the pathogenesis of acute GVHD.38,39 Perturbations of microbiota in mice can exacerbate or ameliorate GVHD through a variety of mechanisms,40,41 but the extent to which alterations in the composition of intestinal microbiota cause acute GVHD or result from acute GVHD in humans remains to be defined. However, preliminary studies showing striking improvement after fecal microbiota transplantation in patients with severe intestinal GVHD have suggested that alterations in the composition of intestinal microbiota may drive persistence of the disease in some patients.42,43 Standardization of products to be tested in future studies and development of methods that accurately identify patients who are likely to respond pose major challenges to further progress. As an alternative approach, preclinical evidence has suggested that R-spondin1 could be used to induce differentiation of intestinal stem cells to Paneth cells that secrete antimicrobial α-defensins, thereby preventing GVHD-mediated dysbiosis.44
Thrombotic microangiopathy
Acute GVHD can cause endothelial injury resulting in thrombotic microangiopathy (TMA).45 High serum concentrations of angiopoietin 246,47 and nitrates48 before HCT make endothelial cells more vulnerable to injury caused by GVHD, thereby contributing to the development of steroid-refractory disease. Preclinical studies have shown that the plasma protein Gas6 contributes to the pathogenesis of TMA associated with GVHD through its interaction with the receptor tyrosine kinase Mer.49 This study also showed that patients with acute GVHD have high serum Gas6 concentrations that correlate with biomarkers of TMA. Taken together, these observations suggest that inhibition of the Gas6-Mer pathway could provide therapeutic benefit in patients with SR-aGVHD complicated by TMA.
Persistent activation of complement via the alternative pathway might contribute to TMA in patients with SR-aGVHD.50 Earlier studies implicated calcineurin inhibitors or sirolimus in the development of TMA.51,52 These observations raise questions of whether TMA associated with GVHD should be treated with complement inhibitors or by withdrawal of calcineurin inhibitors or sirolimus. I would consider the use of complement inhibitors only in cases with unequivocal evidence of complement activation. A retrospective review of cases at our center showed no difference in survival between patients who had calcineurin inhibitors or sirolimus withdrawn vs those who did not.53 Laboratory tests indicating the extent to which these agents contribute to TMA would be extremely helpful in clinical management.
Intestinal viruses
Legoff et al54 characterized the gut virome before and after allogeneic HCT in 44 patients to investigate potential links between changing viral dynamics and the development of intestinal GVHD. In that study, a time-dependent Cox proportional-hazards model showed that detection of picobirnaviruses in the stool predicted the subsequent development of severe intestinal GVHD (hazard ratio, 2.7; 95% confidence interval, 1.5-4.9). That study has opened a new avenue of clinically relevant investigation in the field.
Conclusions
We currently have a wide choice of agents that could be used to treat SR-aGVHD but very little reliable information to determine which ones might be best for any given patient. Many insights regarding the pathogenesis of GVHD have emerged during the past 4 decades, but challenges remain in understanding why the disease is refractory to steroid treatment in some patients. The field would benefit from greater emphasis on well-designed risk-stratified prospective studies, enrolling patients before the disease becomes irreversible, using objective laboratory measures added to clinical measures of response, and focusing on survival as the outcome of greatest importance.
The online version of this article contains a data supplement.
Acknowledgments
The author thanks Keith Loeb for assistance with photomicrographs and for accurate description of pathologic findings, as well as Geoff Hill and Phuong Vo for reviewing the manuscript and providing helpful suggestions.
Authorship
Contribution: P.J.M. wrote the manuscript.
Conflict-of-interest disclosure: P.J.M. has served on advisory boards or consulted for Neovii Biotech GmbH, Genentech, Enlivex Therapeutics, and Pharmacyclics and has received institutional research funding from AbGenomics. P.J.M. provided an invited lecture, sponsored by Janssen, to the 2019 meeting of the Israeli Society of Hematology and Transfusion Medicine; Janssen had no input regarding the content of the lecture. Funding was used solely for travel costs and housing directly related to the meeting; all arrangements were made by a third party, and he did not receive an honorarium.
Correspondence: Paul J. Martin, Fred Hutchinson Cancer Research Center, P.O. Box 19024, 1100 Fairview Ave N, D2-100, Seattle, WA 98109-1024; e-mail: pmartin@fredhutch.org.