Abstract
Cytomegalovirus (CMV) reactivation remains one of the most common and life-threatening infectious complications following allogeneic hematopoietic stem cell transplantation, despite novel diagnostic technologies, several novel prophylactic agents, and further improvements in preemptive therapy and treatment of established CMV disease. Treatment decisions for CMV reactivation are becoming increasingly difficult and must take into account whether the patient has received antiviral prophylaxis, the patient’s individual risk profile for CMV disease, CMV-specific T-cell reconstitution, CMV viral load, and the potential drug resistance detected at the time of initiation of antiviral therapy. Thus, we increasingly use personalized treatment strategies for the recipient of an allograft with CMV reactivation based on prior use of anti-CMV prophylaxis, viral load, the assessment of CMV-specific T-cell immunity, and the molecular assessment of resistance to antiviral drugs.
Introduction
Cytomegalovirus (CMV) is a latent virus that belongs to the family of herpesviruses and is one of the common viral pathogens that can reactivate after hematopoietic stem cell transplant (HCT) during the time of T-cell deficiency or dysfunction. It reactivates in ∼60% to 70% of CMV-seropositive patients, and primary infection affects 20% to 30% of CMV-seronegative recipients transplanted from CMV-seropositive donors. Uncontrolled CMV reactivations can lead to a life-threatening multiorgan CMV disease, such as pneumonia, gastroenteritis, or retinitis.1-5 CMV-seropositive patients undergoing allogeneic HCT (allo-HCT) have an overall higher mortality posttransplant compared with CMV-seronegative patients, especially when undergoing an allograft from an unrelated or mismatched donor.1-9 The risk of prolonged and recurrent reactivation, as well as mortality, may be even higher if the donor is CMV seronegative.8,10-12
A correlation between T-cell deficiency and dysfunction, especially CD4 deficiency and CMV reactivation, has been seen in patients with HIV infection and following solid organ transplantation.13 In HCT patients, a low lymphocyte count and CD4+ T-cell count <50 per microliter at 3 months posttransplant are risk factors for the development of late CMV disease.14 Hakki et al15 also showed that low CD4 T-cell counts (<100 per microliter) and low CD8 T-cell counts (<50 per microliter) at this time point are associated with poor CMV-specific immunity.
Several new developments, such as novel sensitive and rapid diagnostic assays, definition of risk factors, and highly active anti-CMV agents for preemptive therapy, have contributed to reduce the incidence of CMV disease and CMV-related complications posttransplantation8,14,16-25 ; novel agents, such as maribavir, may further expand the armamentarium that is available for preemptive therapy.26
However, CMV reactivation remains one of the most common and life-threatening infectious complications following allo-HCT, and reactivation in the era of routine use of prophylaxis poses novel complexities.5,7,19
In the past 5 years, there have been 2900 publications on the treatment of CMV infection and 70 publications on the management of CMV reactivation following allo-HCT. These numbers pose a challenge to even the most efficient stem cell transplant physician who needs to critically digest the available information, as well as highlights the continued complexities in the management of CMV reactivation following allo-HCT.5,24,27,28
Current strategies to prevent or treat CMV reactivation
The development of highly effective antiviral agents against CMV and the availability of novel and sensitive assays for the detection and quantification of the virus have resulted in the emergence of 2 main strategies to prevent CMV-related outcomes among allograft recipients: antiviral prophylaxis and preemptive therapy based on sensitive detection techniques.
Preemptive antiviral treatment is triggered by early detection of CMV reactivation, before clinical manifestations of CMV disease occur, and it has reduced the incidence of CMV disease.14,17 Preemptive antiviral therapy is based on surveillance by quantitative polymerase chain reaction (PCR) assays, which allows the initiation of preemptive therapy above a certain detection threshold,18,29 depending on the risk of CMV disease in a specific patient.19
The introduction of the international standard has improved the interlaboratory variability of PCR assays,30 but significant differences remain that continue to pose challenges in comparing results between laboratories.31 With this strategy, the risk of early-onset CMV disease (before 100 days posttransplant) is <3%, but patients continue to be at risk for late-onset CMV disease and CMV-related complications,1,21,25,32-36 even in the current era,12 and resistant/refractory CMV infection remains a problem in a significant proportion of patients.5
Anti-CMV prophylaxis
Several studies12,37-41 demonstrated that CMV reactivation posttransplant was associated with an increased risk for overall and all-cause mortality, independent of the use of preemptive therapy. Importantly, the risk increased with increasing viral load.42 Therefore, prevention of viral replication, rather than surveillance-based preemptive therapy, might be beneficial for a seropositive patient following allo-HCT.12,43,44 In the 1980s and 1990s, anti-CMV prophylaxis with high-dose acyclovir or valacyclovir was studied and shown to have some effect on CMV reactivation45,46 ; however, it is not widely used because its efficacy in preventing CMV disease is limited.47
For decades, highly effective agents to control CMV infection have been limited to drugs with significant toxicity (ie, ganciclovir, foscarnet, and cidofovir). Ganciclovir is associated with hematotoxicity and thus, an increased incidence of secondary bacterial and fungal infections,48-52 IV foscarnet is associated with electrolyte disturbances and severe nephrotoxicity,53-56 and cidofovir is associated with nephrotoxicity, hematotoxicity, and ocular toxicity.45 Ganciclovir is the only drug that has been evaluated as prophylaxis in randomized trials14,49,57 ; however, it did not result in improved overall survival because of severe neutropenia and secondary bacterial and fungal infections.58,59
The results from relatively small uncontrolled trials provide support for prophylaxis with valganciclovir or foscarnet only in very high-risk patients.60,61
Valganciclovir prophylaxis was not shown to provide improved protection from CMV disease compared with PCR-guided preemptive therapy for the prevention of late-onset CMV disease.62 Thus, until recently, most centers have not used antiviral prophylaxis; instead, they have relied on preemptive therapy as the management strategy.47 This has changed recently with the introduction of letermovir.
Letermovir prophylaxis
Letermovir inhibits the human CMV (HCMV) terminase complex, which is a novel mechanism of action. It was studied in 2 randomized placebo-controlled studies for CMV prophylaxis, including in 686 patients following allo-HCT.21,22 The introduction of letermovir is an important advance because it is not myelotoxic or nephrotoxic, and does not require dose adjustments based on renal and mild to moderate hepatic dysfunction.63 Furthermore, letermovir is available in oral and IV formulations, allowing early administration posttransplant and during phases when patients are acutely ill or unable to take oral medication. Dose adjustments are required in patients receiving graft-versus-host disease (GvHD) prophylaxis with cyclosporine.63,64
In a phase 3 trial, prophylaxis with letermovir significantly reduced the rate of clinically significant CMV infection defined as development of CMV disease or the need for administration of preemptive anti-CMV therapy. In addition, all-cause mortality was reduced by week 24 following allo-HCT; however, statistical significance was lost by week 48. The reduction in clinically significant CMV infection and all-cause mortality was especially pronounced in patients at high risk for CMV reactivation (eg, patients undergoing an HCT from a haploidentical or mismatched donor or those receiving anti-thymocyte globulin [ATG] for GvHD prophylaxis).21,22 In a post hoc analysis, it was shown that letermovir also reduced all-cause mortality at week 48 after allo-HCT in patients who developed clinically significant CMV infection.3 Since its approval, there has been increased use of letermovir prophylaxis in allograft recipients posttransplant, especially in patients at high risk for CMV reactivation and disease. The results of the phase 3 trial, as well as clinical postlicensing experience, indicate that the use of letermovir delays viral reactivation to the time after discontinuation of prophylaxis in patients with continued immunosuppression. The impact of this late reactivation, how patients at risk are best identified, and how it should be best managed are being studied.
Unfortunately, 2 other novel antiviral agents (maribavir, brincidofovir) and a DNA vaccine (ASP113) failed to improve the CMV-related outcomes in phase 3 prophylaxis trials,25,32,65 although phase 2 trials of the DNA vaccine,66 maribavir,67 and brincidofovir68 had demonstrated significantly fewer CMV events and lower antiviral activity. Early trials of a CMV peptide and modified vaccinia Ankara vaccines showed promising results in initial randomized trials69,70 and are being evaluated further.
Case 1
A 37-year-old woman was diagnosed with a high-risk multiple myeloma [t(t4;14) and 17p del]; following 4 cycles of induction therapy and tandem high-dose melphalan therapy with autologous HCT, she exhibited progressive myeloma at 9 months posttransplant. A male CMV-seronegative donor with a 9/10 HLA match was identified for this CMV-seropositive patient. Following reinduction and conditioning therapy with fludarabine/treosulfan plus ATG and a peripheral blood stem cell transplant, she received cyclosporine and methotrexate for GvHD prophylaxis. Antiviral prophylaxis in this high-risk patient included acyclovir prophylaxis for herpes simplex virus (HSV)/varicella zoster virus (VZV) infection and letermovir (240 mg/d; reduced dose because the patient received cyclosporine) beginning on day +9. She was monitored weekly for CMV reactivation but did not experience a documented CMV infection. The patient achieved a partial response for her multiple myeloma, and donor lymphocyte infusion plus lenalidomide was started. The patient stopped letermovir prophylaxis on day +80 when anti-myeloma therapy was started. The ongoing viral screening revealed CMV reactivation on day +94 with a viral load of 18 900 IU/mL. Because letermovir prophylaxis has not been shown to induce cross-resistance to other anti-CMV agents in a patient with full hematopoietic reconstitution, we started preemptive therapy with valganciclovir in an outpatient setting. After 4 weeks of antiviral therapy, the patient finally cleared the CMV infection (Figure 1).
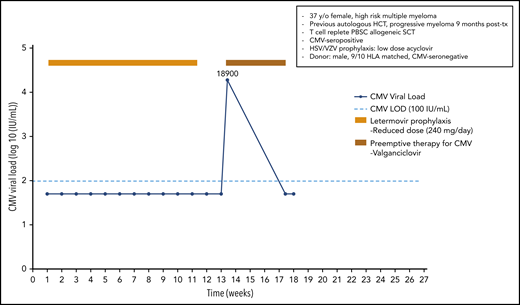
Case 1: engraftment, viral reactivation, antiviral prophylaxis, treatment, and other relevant clinical data of a 37-year-old woman with high-risk multiple myeloma.
Case 1: engraftment, viral reactivation, antiviral prophylaxis, treatment, and other relevant clinical data of a 37-year-old woman with high-risk multiple myeloma.
An allograft recipient at high-risk for CMV disease benefits the most from antiviral prophylaxis. In the phase 3 trial of CMV prophylaxis with letermovir,22 very few patients developed CMV reactivation with quantifiable viral load; among those was 1 patient with a mutation (UL56 V236M) that confers letermovir resistance. Despite the fact that only a few breakthrough infections during prophylaxis with letermovir were reported, we monitor patients on letermovir prophylaxis for CMV reactivation. However, it should be recognized that, probably as a result of the mechanism of action of letermovir, low-grade DNAemia does not indicate letermovir failure. Rapid breakthrough infection was reported in some patients who received letermovir for secondary prophylaxis or when used for treatment of CMV reactivation with high viral loads.71 Mutations in codons 231 to 369 of the UL56 gene have been described in these patients with letermovir-resistant CMV infection.72 All patients who developed CMV reactivation during or following letermovir prophylaxis responded to preemptive antiviral therapy and did not demonstrate induction of cross-resistance to other antiviral drugs following letermovir prophylaxis.
Case 2
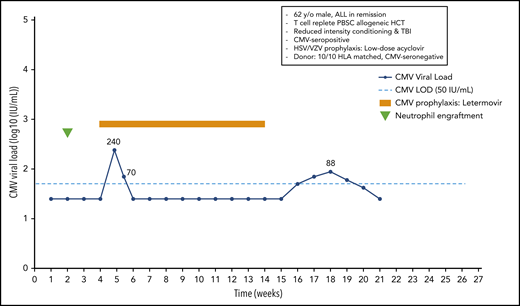
A 62-year-old man with acute lymphoblastic leukemia in remission received a 10/10 HLA-matched T-cell–replete peripheral blood stem cell transplant following reduced-intensity conditioning with melphalan, fludarabine, and total-body irradiation (300 cGy). The patient was CMV seropositive, and the donor was seronegative. He received low-dose acyclovir for prevention of HSV and VZV. Neutrophil engraftment occurred at day 14, and oral letermovir was started on day 28. Weekly PCR surveillance was performed. On day 34 after HCT, the patient showed a CMV plasma viral load of 240 IU/mL. Because the patient did not have acute GvHD, and thus, did not receive systemic steroids, letermovir prophylaxis was continued. The viral load was 70 IU/mL on day 38, and subsequent weekly viral load tests were negative until day 100, when letermovir was discontinued. Subsequent weekly PCR surveillance showed several low-level viral load results ranging from 42 to 88 IU/mL between days 112 and 140, which were not treated (Figure 2).
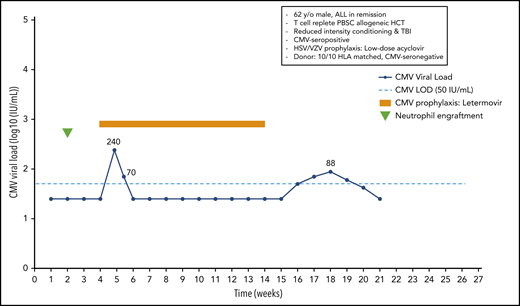
Case 2: engraftment, viral reactivation, antiviral prophylaxis, treatment, and other relevant clinical data of a 62-year-old man with ALL in remission.
Case 2: engraftment, viral reactivation, antiviral prophylaxis, treatment, and other relevant clinical data of a 62-year-old man with ALL in remission.
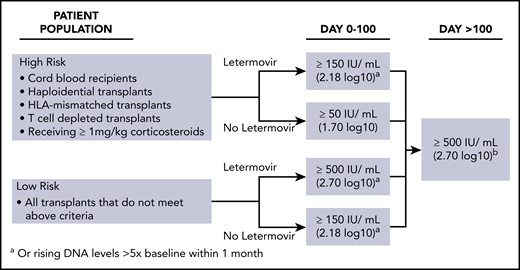
Subclinical reactivation on antiviral prophylaxis is not uncommon. Indeed, several studies performed with ganciclovir and valganciclovir prophylaxis showed subclinical reactivation rates of 20% to 40%.15 Most episodes resolved without changing the antiviral prophylaxis regimen. Similar results have recently been observed with letermovir prophylaxis. We start letermovir in low-risk patients after neutrophil engraftment and when they can take oral medication, but no later than day 28. We continue weekly PCR surveillance in letermovir recipients and treat breakthrough infections at levels similar to those used in the phase 3 randomized controlled trial22 (Figure 3). The patient illustrates that low-level reactivation can resolve on continued letermovir prophylaxis without further intervention in a low-risk situation. We would have treated a viral load of 220 IU/mL if the patient had received corticosteroids at a dose ≥1 mg/kg or met other high-risk criteria (Figure 3). Earlier data from the ganciclovir era showed that subclinical reactivation that occurred during prophylaxis improved CMV-specific T-cell immune reconstitution.15 Whether such an effect also occurs in recipients of letermovir prophylaxis is being studied. Our patients had a few episodes of low-level reactivation after discontinuation of letermovir at day 100, which did not require preemptive treatment.

Viral load thresholds for starting preemptive therapy. Adapted from CMV Prevention: Prophylaxis, Surveillance, and Preemptive Therapy in Hematopoietic Stem Cell Transplant Recipients Guidelines.111
Viral load thresholds for starting preemptive therapy. Adapted from CMV Prevention: Prophylaxis, Surveillance, and Preemptive Therapy in Hematopoietic Stem Cell Transplant Recipients Guidelines.111
Because antiviral prophylaxis, especially with letermovir, is not yet available at all centers performing allo-HCT and is often not used in low-risk patients, we discuss the management of patients after allo-HCT who received prophylaxis with valganciclovir or those who did not receive antiviral prophylaxis.
Case 3
A 52-year-old woman HCMV-seropositive patient with a Philadelphia-positive B-cell acute lymphoblastic leukemia entered a complete remission following induction therapy and received a 9/10 matched cord blood transplant. Because this patient is at extremely high risk for CMV disease, she received oral valganciclovir prophylaxis beginning on day 24 posttransplant (starting with 2 × 900 mg/d, which was reduced to 2 × 450 mg/d because of poor graft function). This strategy had been shown to be effective in previous studies in high-risk patients.14,60 On day 74, the patient developed a CMV reactivation with a viral load of 13 650 IU/mL. The patient was switched to IV foscarnet (2 × 60 mg/kg of body weight), which was stopped after 14 days because of impaired renal function. Resistance testing revealed a C592G UL97 mutation not associated with high-level resistance to ganciclovir; thus, IV ganciclovir was restarted at full dose, and a CMV-specific T-cell product from a 9/10 HLA-matched CMV-seropositive third-party donor was generated using the cytokine catch assay and transfused to the patient. The patient had cleared the viral infection 14 days later, ganciclovir therapy was stopped, and the weekly blood samples obtained from the patient remained negative for CMV in all subsequent analyses (Figure 4).

Case 3: engraftment, viral reactivation, antiviral prophylaxis, treatment, and other relevant clinical data of a 52-year-old woman with Philadelphia-positive ALL in CR.
Case 3: engraftment, viral reactivation, antiviral prophylaxis, treatment, and other relevant clinical data of a 52-year-old woman with Philadelphia-positive ALL in CR.
When CMV reactivation with a high viral load is detected following long-term antiviral prophylaxis or preemptive therapy with valganciclovir (>6 weeks of antiviral drug exposure) in a patient who carries a high risk for CMV disease, a drug-resistant CMV infection must be suspected.5,73-77 Additionally, the reduction in the dose of valganciclovir might have further increased the risk of ganciclovir resistance. The bioavailability of valganciclovir is variable.78
Therefore, therapeutic drug monitoring is used routinely in Stockholm to avoid underdosing, as well as overdosing.79 In addition, dose reductions in (val)ganciclovir because of hematotoxicity should be avoided; the full dose of the drug should be maintained by adding growth factor support.27,80,81 Because resistance testing revealed a C592G UL97 mutation, which is associated only with low-level resistance, the patient was retreated with high-dose IV ganciclovir when the renal function deteriorated during foscarnet therapy. Because the patient had impaired renal function, which would potentially limit the use of all available antiviral agents at that time, as well as had an infection with already low-level ganciclovir resistance, we decided to restart therapy with IV ganciclovir and to add a CMV-specific T-cell product from a CMV-seropositive third-party donor (the stem cell donor was CMV seronegative). Transfer of CMV-specific T-cell products containing CMV-specific CD4+ and CD8+ T cells selected from CMV-seropositive donors by cytokine catch assay has been demonstrated to be safe and successful in controlling drug-resistant CMV infection in a small number of CMV-seropositive recipients of a cord blood transplant,82-86 whereas infusions of CMV-specific T-cell products from third-party donors containing highly purified CD8+ CMV-specific T cells selected by streptamer technology failed to clear drug-resistant CMV infection.87 An alternative approach in this patient (that would have been done in Seattle) is to use high-dose ganciclovir (7.5-10 mg/kg twice daily, adjusted for renal function) with granulocyte colony-stimulating factor (G-CSF) support, given the low-level resistance mutation.24,76,77 Other successful alternative pharmacological therapies for allograft recipients with resistant and refractory CMV infection include artesunate and leflunomide, which were reported in small patient cohorts.8,24,88-92 The most interesting new pharmacological treatment for refractory CMV infection is maribavir; in doses ≥400 mg twice daily, it was shown to be active against refractory and resistant CMV infection in a large randomized double-blind phase 2 trial in patients following hematopoietic or solid organ transplantation.93
The role of letermovir in routine clinical use will have to be established. It has not yet been introduced in all countries; because of cost issues, some centers have decided only to give it to selected primarily high-risk patients. No data exist regarding letermovir use in children, and the optimal duration of prophylaxis also needs to be properly studied. Therefore, we will also discuss the management of patients with CMV reactivation not receiving antiviral prophylaxis.
Case 4
A 55-year-old HCMV-seropositive man was diagnosed with acute myeloid leukemia and achieved a complete response following 2 cycles of induction therapy. An HLA-identical HCMV-seropositive brother was identified as a donor, and an allo-HCT was performed following reduced-intensity conditioning. The patient received acyclovir prophylaxis for HSV/VZV infection. No anti-CMV prophylaxis was introduced. On day +20 the patient developed grade 1 skin GvHD, which resolved without any treatment. The patient underwent weekly monitoring using quantitative real-time PCR. In addition, CMV-specific T-cell numbers were monitored every 2 weeks, using a home-made streptamer assay, as part of a clinical study. At day 90 posttransplant, a CMV load of 1280 IU/mL was noted, and immune monitoring at that time revealed the presence of CD8+ CMV-specific T cells in the peripheral blood, as determined by streptamer assay.
With the rather low viral load in a patient at low risk for CMV disease (CMV-seropositive HLA-identical sibling donor, no severe GvHD, no ATG used for conditioning) with a documented CMV-specific T-cell response, we decided not to use preemptive treatment; instead, we continued to monitor viral load and CMV-specific T-cell responses. The next 2 analyses showed a decrease in the viral load to 840 IU/mL and then to 472.50 IU/mL and a persisting CMV-specific T-cell response. Without any anti-CMV therapy, the patient finally cleared the CMV infection, and the continued monitoring (routinely performed at the Würzburg Center until day 180 in a patient with a CMV reactivation early posttransplant) did not reveal any further CMV reactivations (Figure 5).
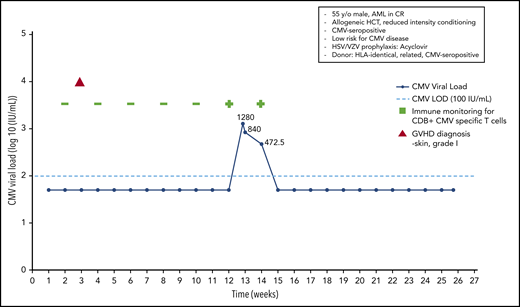
Case 4: engraftment, viral reactivation, antiviral prophylaxis, treatment, and other relevant clinical data of a 55-year-old man with AML in CR.
Case 4: engraftment, viral reactivation, antiviral prophylaxis, treatment, and other relevant clinical data of a 55-year-old man with AML in CR.
Monitoring of CD8+ and/or CD4+ CMV-specific T cells can be performed using different techniques (eg, streptamers, pentamers, and other MHC multimers) or CMV-specific (more recently CMV-specific peptide) T-cell responses (cytotoxic activity against CMV-infected targets, T-cell proliferation, cytokine production [eg, by enzyme-linked immunospot assay or flow cytometry]).15,94-96 Commercial tests are now also available to monitor CMV-specific and even polyfunctional (interferon-γ, tumor necrosis factor-α) CMV-specific T cells following allo-HCT.97-105 If a patient has a documented CMV-specific T-cell response at the time of detection of virus reactivation with a low or medium viral load, we continue monitoring the viral load and delay antiviral chemotherapy until the viral load increases or the CMV-specific T-cell response diminishes or completely disappears.5,106
In patients at low risk for CMV disease (R+/D+, donor HLA-identical sibling, no severe GvHD) with a CMV reactivation and a low to medium viral load, we continue monitoring for an increase in viral load but do not start preemptive therapy.107-109 In Würzburg, we believe that monitoring for CMV-specific T-cell responses can further inform treatment decisions95,105 ; thus, we believe the documented CMV-specific CD8+ T-cell response further reduced the risk for the development of CMV disease in this patient. This strategy has not been tested in a randomized trial, and we have to caution that, in rare cases at the Fred Hutchinson Cancer Research Center, CMV disease has been seen in patients with a documented CMV-directed T-cell response, indicating that not all of the T-cell responses detected are protective.
Case 5
A 28-year-old man was diagnosed with high-risk acute myeloid leukemia. Following 2 cycles of induction therapy he entered a complete remission and, following myeloablative conditioning, received a transplant from a fully matched unrelated donor. GvHD prophylaxis included ATG and cyclosporine/methotrexate. The patient was CMV seropositive and received a transplant from a CMV-seropositive donor. Acyclovir prophylaxis for HSV/VZV infection was administered. On day +26, the patient developed grade 3 acute GvHD involving the skin and the intestinal tract. The patient received high-dose corticosteroid therapy (methylprednisolone, 2 mg/kg of body weight) and additional ruxolitinib for insufficient control of intestinal GvHD following tapering of the steroids.
On day 48, viral load monitoring showed a high CMV viral load (35 700 IU/mL) and, because of poor marrow function, the patient received foscarnet (2 × 60 mg/kg of body weight). Following 2 weeks of therapy the viral load was reduced to 12 600 IU/mL, and foscarnet therapy was continued. Because of deterioration of renal function, ganciclovir (2 × 5 mg/kg) was started, with 1 week of G-CSF for ongoing poor marrow function. With further decreasing viral load and improvement of intestinal GvHD, the patient was discharged and therapy was switched to oral valganciclovir until CMV infection was cleared. In Stockholm, the treatment would have been guided by therapeutic drug monitoring of ganciclovir, and the dose would have been adjusted accordingly. Monitoring for CMV infection was continued weekly until day +180. Another episode of CMV reactivation required treatment with valganciclovir for another 14 days until clearance of the virus infection (Figure 6).

Case 5: engraftment, viral reactivation, antiviral prophylaxis, treatment, and other relevant clinical data of a 28-year-old man with high-risk AML in CR.
Case 5: engraftment, viral reactivation, antiviral prophylaxis, treatment, and other relevant clinical data of a 28-year-old man with high-risk AML in CR.
Although no formal comparative study between IV ganciclovir and valganciclovir has been performed, many centers routinely use valganciclovir as first-line preemptive therapy. For patients not able to take an oral drug, IV ganciclovir or foscarnet is commonly used. A clinical trial performed by the Infectious Disease Working Party of the European Blood and Marrow Transplantation Society56 showed similar efficacy, but different toxicities, for preemptive therapy using IV ganciclovir or foscarnet. Thus, in patients with poor marrow function, we prefer preemptive therapy with foscarnet, whereas in patients with deterioration of renal function we would switch to IV ganciclovir. If both neutropenia and renal insufficiency are present, one can consider ganciclovir or valganciclovir with preemptive G-CSF support.80,81 We tend to administer preemptive therapy for CMV infection until viral clearance for ≥14 days14,44,110 in patients failing both ganciclovir and foscarnet because of poor efficacy or unacceptable toxicity. Cidofovir is 1 possible option.
Conclusions
The increased use of anti-CMV prophylaxis, quantitative PCR assays, assays to define the CMV-specific T-cell response posttransplant, and novel techniques to detect resistance to antiviral drugs, as well as a better understanding of risk factors for CMV disease, make the identification of the optimal therapy for a patient with CMV reactivation following allo-HCT challenging. Therefore, we increasingly use personalized treatment strategies when we approach allograft recipients with a documented CMV reactivation.
We consider the following factors when making decisions about how to treat CMV reactivation in a patient who underwent allo-HCT:
Did the patient receive anti-CMV prophylaxis when CMV reactivation was detected or prior to the reactivation? If so, which agent and for how long?
Did high-level CMV reactivation occur during/following antiviral prophylaxis? If so, screening for antiviral resistance might be indicated.
Is the patient at high risk for CMV disease (cord blood transplant, haploidentical HCT, ATG for seroprophylaxis, graft from an HLA-mismatched unrelated donor)?
CMV load/kinetics
CMV-specific T-cell reconstitution
Comorbidities (eg, foscarnet for patients with poor marrow function or ganciclovir for patients with impaired renal function)
The treatment strategies used in our patients (Table 1) are based on these considerations; however, not all of them have been confirmed in randomized controlled trials. For instance, although CMV-specific T-cell immunity correlates with protection from high-level reactivation in observational studies, our strategy to use it to withhold preemptive therapy (Case 3) should be systematically evaluated, optimally in a randomized trial. Also, the viral load thresholds that we used to start preemptive therapy while on letermovir prophylaxis (Figure 3) have been developed by synthesizing the data on subclinical reactivation in recipients of antiviral prophylaxis with letermovir and ganciclovir/valganciclovir,20,74 but they may need to be adjusted as more experience with letermovir emerges.
Acknowledgments
The authors thank Rachel Blazevic for assistance with the figures.
This work was supported by Deutsche Forschungsgemeinschaft with the research group FOR 2830, Advanced Concepts in Cellular Immune Control of Cytomegalovirus (project P09; H.E.), and the Collaborative Research Centre (Transregio CRC 221, A03). M.B. was supported by the National Institutes of Health (NIH) National Heart, Lung, and Blood Institute (K24HL093294-02) and the NIH National Institute of Allergy and Infectious Diseases (AI-2014028).
Authorship
Contribution: All authors submitted case presentations and discussions, and were involved in the writing of the manuscript.
Conflict-of-interest disclosure: P.L. reports personal fees from AiCuris and grants from Astellas, Oxford Immunotech, Takeda (Shire), and MSD outside of the submitted work. M.B. reports grants and personal fees from Merck, Takeda/Shire, Gilead Sciences, and VirBio; grants from Astellas and Chimerix; personal fees, including options to acquire equity from Helocyte and EvrysBio; and personal fees from GlaxoSmithKline, Moderna, and AlloVir, outside of the submitted work. H.E. declares no competing financial interests.
Correspondence: Hermann Einsele, Department of Internal Medicine II, University Hospital Würzburg, Oberdürrbacher Str 6, 97080 Würzburg, Germany; e-mail: einsele_h@ukw.de.