

Introduction: Myelofibrosis (MF) is a chronic myeloproliferative neoplasm characterized by hyper-activation of the JAK-STAT pathway. More than half of the patients carries the JAK2V617F mutation. Cytokine overproduction, which is the hallmark of MF, is driven by multiple signaling pathways (NF-KB and MAPK) beyond JAK-STAT and is reduced but not abrogated by Ruxolitinib (RUX). RUX, which is the only JAK1/2 inhibitor approved for the treatment of splenomegaly and symptoms associated with MF, suppresses myeloproliferation (JAK2-driven) and release of pro-inflammatory cytokines (JAK1-driven). Microvesicles (MVs), which have a role in the inflammatory network and are critical players in the regulation of immunity through cytokine signaling, are released from a broad variety of cells with effects on communication among cells. Infections are one of the main causes of morbidity and mortality in MF. The increased infectious risk is thought to arise from dysfunction of key immune cells, including T, natural killer and dendritic cells, which further aggravates after JAK1/2 inhibition therapy. In this scenario, even though MF monocytes (Mo) are over-activated and show inflammatory features, their contribution still needs to be clarified.
Aims: To study the role of circulating Mo in the inflammatory network of MF and to investigate whether and to what extent in vivoJAK1/2 inhibition may affects their in vitro cytokine producing ability.
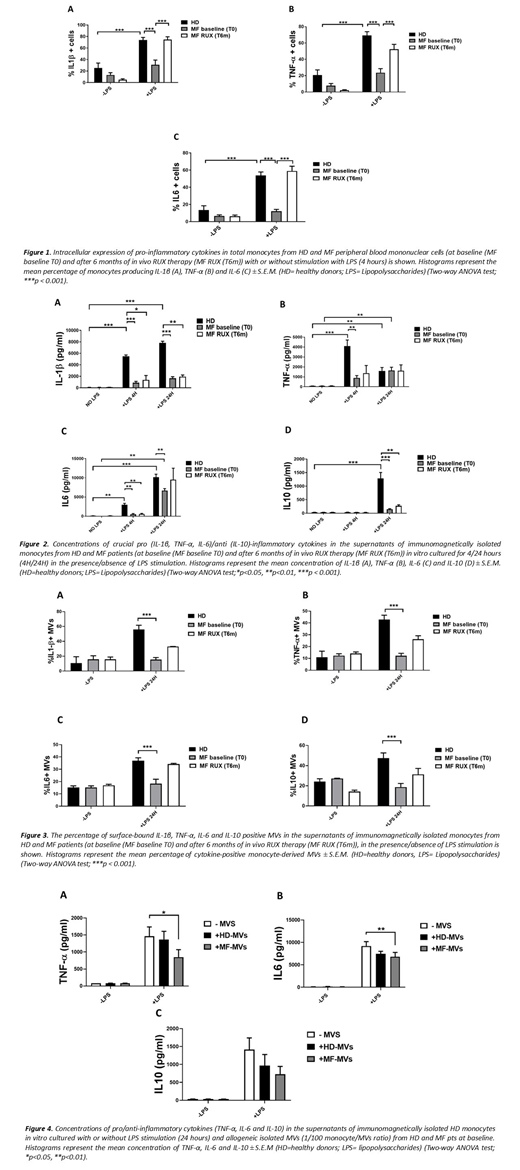
Methods: EDTA-anticoagulated peripheral blood was collected from 12 JAK2V617F mutatedMF patients before (Baseline) and after 6 months of RUX therapy and from 10 age/sex-matched healthy donors (HD). After 4 hours in vitro stimulation of mononuclear cells (PBMCs) with lipopolysaccharides (LPS) and in the presence of brefeldin A, the Interleukin (IL)-1β, -6, -10 and Tumor Necrosis Factor (TNF)-α producing Mo (CD14+ cells) were measured by intracellular staining and flow cytometry analysis. In parallel experiments, upon LPS stimulation, free and MVs-bound cytokines (IL-1β, IL-6, IL-10 and TNF-α) have been measured in the supernatants of immunomagnetically isolated HD/MF-Mo by flow cytometry. In addition, after isolation with ultracentrifugation from platelet poor plasma, titrating doses of circulating HD/MF MVs were co-cultured for 24 hours with immunomagnetically isolated HD-Mo and, upon LPS stimulation, inflammatory cytokines secretion was analysed in the supernatants by flow cytometry.
Results: To characterize the cytokine producing ability of Mo we analyzed the IL1-β, TNF-α, IL-6 and IL-10 positive MF/HD Mo in response to LPS stimulation. At baseline, the percentages of MF-Mo producing pro-inflammatory cytokines (IL-1β, IL-6, TNF-α) after 4 hours LPS stimulation was highly reduced as compared with the HD counterparts (Fig. 1). No IL-10-positive cells were detected with LPS stimulation (data not shown). To confirm the data, we analysed the free and MVs-bound cytokines in the culture supernatants upon LPS stimulation. At baseline, MF-Mo showed defective capacity to secrete free (Fig.2) and MVs-bound (Fig.3) IL-1β, IL-6, TNF-α and IL-10. Interestingly, the isolated circulating MF-MVs inhibited the LPS-driven inflammatory cytokines in vitro secretion of HD-Mo (Fig.4). Six months-RUX therapy reactivated the in vitro MF-Mo ability to produce intracellular inflammatory cytokines (Fig.1) and to secrete MVs-bound inflammatory cytokines in response to LPS stimulation (Fig.3). Conversely, the MF-Mo ability to secrete free cytokines in the supernatants in response to LPS stimulus remained lower than the HD counterparts (Fig.2).
Conclusions: These data further refine the immune dysfunction of MF by demonstrating defective cytokine production/secretion of circulating Mo in response to infectious stimulus (LPS). This defect might be due, at least in part, to the inhibitory activity of circulating MF-MVs. Importantly, in vivoJAK1/2 inhibition ameliorates Mo cytokines production and promotes the MVs-based inflammatory cytokine signaling, suggesting that the increased infectious risk of MF patients undergoing RUX therapy is not due to defective inflammatory signals of circulating Mo. These findings contribute to better interpreting the off-target efficacy of JAK1/2 inhibition and to envisaging strategies aimed at facilitating the immune surveillance in MF.
Martinelli:BMS: Consultancy; Pfizer: Consultancy; ARIAD: Consultancy; Novartis: Consultancy; Roche: Consultancy. Cavo:janssen: Consultancy, Honoraria, Membership on an entity's Board of Directors or advisory committees, Other: travel accommodations, Speakers Bureau; celgene: Consultancy, Honoraria, Membership on an entity's Board of Directors or advisory committees, Other: travel accommodations, Speakers Bureau; AbbVie: Consultancy, Honoraria, Membership on an entity's Board of Directors or advisory committees; amgen: Consultancy, Honoraria, Membership on an entity's Board of Directors or advisory committees, Speakers Bureau; takeda: Honoraria, Membership on an entity's Board of Directors or advisory committees, Speakers Bureau; novartis: Honoraria; sanofi: Honoraria, Membership on an entity's Board of Directors or advisory committees, Speakers Bureau; bms: Honoraria, Membership on an entity's Board of Directors or advisory committees, Speakers Bureau. Palandri:Novartis: Consultancy, Honoraria.

