

Key Points
Plg-RKT facilitates migration of proinflammatory monocyte and macrophage subsets.

Abstract
Membrane-bound plasmin is used by immune cells to degrade extracellular matrices, which facilitates migration. The plasminogen receptor Plg-RKT is expressed by immune cells, including monocytes and macrophages. Among monocytes and macrophages, distinct subsets can be distinguished based on cell surface markers and pathophysiological function. We investigated expression of Plg-RKT by monocyte and macrophage subsets and whether potential differential expression might have functional consequences for cell migration. Proinflammatory CD14++CD16+ human monocytes and Ly6Chigh mouse monocytes expressed the highest levels of Plg-RKT and bound significantly more plasminogen compared with the other respective subsets. Proinflammatory human macrophages, generated by polarization with lipopolysaccharide and interferon-γ, showed significantly higher expression of Plg-RKT compared with alternatively activated macrophages, polarized with interleukin-4 and interleukin-13. Directional migration of proinflammatory monocytes was plasmin dependent and was abolished by anti–Plg-RKT monoclonal antibody, ε-amino-caproic acid, aprotinin, and the aminoterminal fragment of urokinase-type plasminogen activator. In an in vivo peritonitis model, significantly less Ly6Chigh monocyte recruitment was observed in Plg-RKT−/− compared with Plg-RKT+/+ mice. Immunohistochemical analysis of human carotid plaques and adipose tissue showed that proinflammatory macrophages also exhibited high levels of Plg-RKT in vivo. Our data demonstrate higher expression of Plg-RKT on proinflammatory monocyte and macrophage subsets that impacts their migratory capacity.
Introduction
The zymogen plasminogen is activated by urokinase-type plasminogen activator (uPA) or tissue-type PA to the broad-spectrum protease, plasmin. Plasmin is the major enzyme responsible for fibrin degradation in vivo.1,2 In addition, plasmin regulates extracellular proteolysis, by degrading extracellular matrix components and by activating promatrix metalloproteinases.3 Consequently, plasmin is critically involved in physiological and pathological processes requiring cell migration.4
Plasminogen activation is markedly enhanced when plasminogen is bound to cell surfaces, and plasmin is then protected from inactivation by α2-antiplasmin.5 The novel plasminogen receptor, Plg-RKT, is expressed in membranes of a variety of cells and is colocalized with the uPA receptor, uPAR.6 Monocytes and macrophages express Plg-RKT, and this receptor plays a critical role in plasmin-mediated cell migration, invasion, and recruitment during inflammation.7,8
Human monocytes can be divided into functionally distinct subsets, based on expression of CD14 and CD16, into classical monocytes (CMs; CD14++CD16−), intermediate monocytes (IMs; CD14++CD16+), and nonclassical monocytes (NCMs; CD14+CD16++).9 The CD16+ subsets are associated with pathologies characterized by a chronic inflammatory state, including coronary artery disease and obesity.10-12 Macrophages also exhibit distinct functional heterogeneity and plasticity.13 Exposure to interferon-γ (IFN-γ) and lipopolysaccharide (LPS) primes macrophages toward a proinflammatory phenotype, whereas macrophages exposed to interleukin-4 (IL-4) and IL-13 are linked to tissue repair processes.14
We here investigated whether monocyte and macrophage subsets differentially express Plg-RKT and if potential differential expression would have functional consequences for their plasmin-mediated migratory capacity.
Study design
IL-6 and IL-10 were determined by specific enzyme-linked immunosorbent assays according to the manufacturer’s instructions (Abcam, Cambridge, UK).
Studies were approved by the institutions’ ethics committee and performed according to the Declaration of Helsinki. Informed consent was obtained from all participants.
Results and discussion
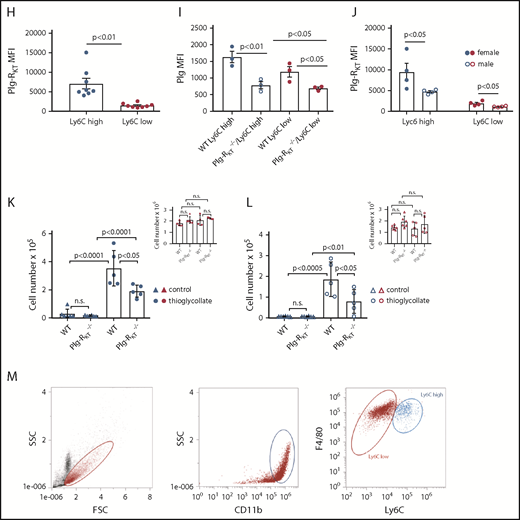
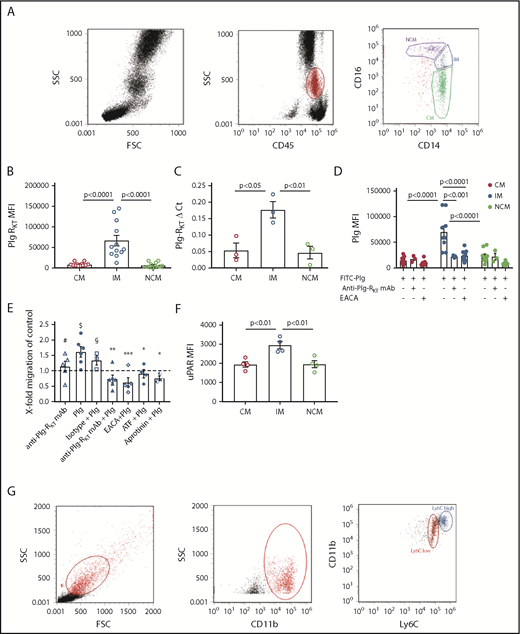
Three subsets of human monocytes can be distinguished, namely CD14++CD16− CMs, CD14++CD16+ IMs, and CD14+CD16++ NCMs (Figure 1A).9 CD16+ subsets are associated with pathologies characterized by a chronic inflammatory state, such as coronary artery disease, obesity, arthritis, inflammatory diseases of the intestinal tract, or systemic lupus erythematosus.10-12 We have shown that in humans with mild inflammation, the CD16+ subsets exhibit the highest inflammatory potential.15 Here, we found that IMs expressed significantly higher levels of cell surface Plg-RKT compared with CMs and NCMs (Figure 1B). This was paralleled in quantitative polymerase chain reaction (Figure 1C). There was a trend for higher Plg-RKT expression on female monocyte subsets (data not shown). IMs specifically bound more fluorescein isothiocyanate (FITC)-plasminogen than either CMs or NCMs. Plasminogen binding was blocked by ε-aminocaproic acid (EACA) and by anti–Plg-RKT monoclonal antibody (mAb)6,7 on IMs (Figure 1D). Furthermore, IM exhibited plasminogen- and plasmin-dependent directed cell migration that was abolished in the presence of either EACA, aprotinin, the aminoterminal fragment of uPA,16 or anti–Plg-RKT mAb, but not by isotype control antibody. The anti–Plg-RKT mAb had no effect on migration in the absence of plasminogen (Figure 1E). IMs expressed significantly higher levels of uPAR (Figure 1F), consistent with uPAR colocalization with Plg-RKT,6 and with the published key role of uPAR in cell migration.17
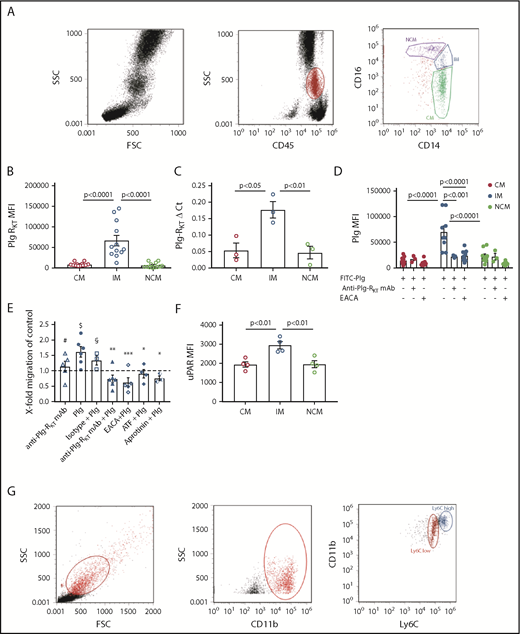
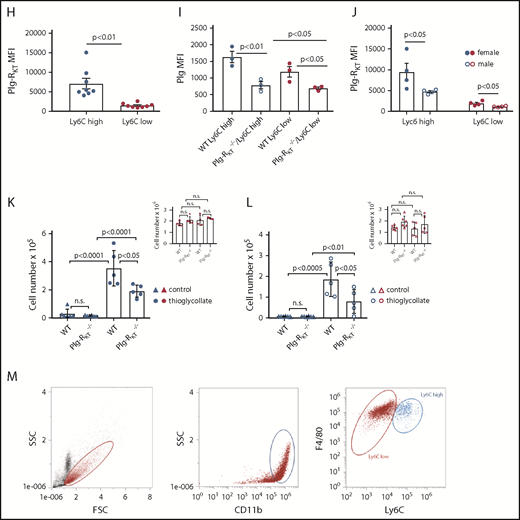
Monocyte subsets and Plg-RKT. (A) Peripheral blood mononuclear cells were isolated from leukapheresis chambers obtained from healthy thrombocyte donors using lymphocyte separation medium 1077 (PromoCell, Heidelberg, Germany) and used for either monocyte subset isolation employing anti-CD45, anti-CD14, and anti-CD16 antibodies or for generating macrophages as described.15,26 Gating strategy for human monocyte subsets in whole blood is shown. The relative amounts of cells per subset were in accordance with published data, CM: 76% ± 5%, IM: 5% ± 2%, NCM: 19% ± 7%.15 (B) Mean fluorescent intensities (MFIs) of Plg-RKT on human monocyte subsets (n = 12). Monocytes in freshly drawn blood were stained with anti-CD14 antibody (PerCP/Cy5.5, Clone HCD14), anti-CD16 antibody (APC-Cy7, Clone 3G8), anti-CD45 antibody (Pacific Blue, Clone HI30), and anti–Plg-RKT mouse monoclonal mAb7H1, prepared and characterized in our laboratory.6,7 Samples were analyzed with a Novocyte Flow cytometer (ACEA Biosciences, San Diego, CA). (C) Plg-RKT mRNA levels in human monocyte subsets. Cells were sorted based on CD14 and CD16 expression into CMs, IM, and NCMs using a BD FACSAria (BD Biosciences, Franklin Lakes, NJ), and afterward, RNA was isolated for quantitative polymerase chain reaction. Primer sequences for Plg-RKT were “tggaacccttttagaaagaatga” and “ttggcagctgcaatttactc” (Universal Probe Library, #73; Roche, Basel, Switzerland). Glyceraldehyde-3-phosphate dehydrogenase was used for normalization (n = 3). (D) Plasminogen binding to human monocyte subsets in the presence or absence of anti–Plg-RKT mAb (clone7H1, developed and characterized in our laboratory)6,7 or EACA. PBMCs were stained with fluorochrome-labeled antibodies against CD45, CD14, and CD16 and incubated with FITC-labeled plasminogen (0.5 µM; n = 9).6 Fluorescence was measured with a Novocyte Flow cytometer. To test specificity of plasminogen binding, samples were additionally incubated with EACA (0.2 M; Sigma Aldrich, St. Louis, MO; n = 9) or with anti–Plg-RKT mAb (140 nM; n = 3). (E) Cell migration of human monocyte subsets in the presence or absence of plasminogen. Sorted IMs were seeded into inserts of Costar Transwell Permeable Supports (5.0-µm pore size; Corning, New York, NY) in RPMI 1640 containing 1% bovine serum albumin (both from Sigma Aldrich). Cells were incubated in total for 1 hour at 37°C, 5% CO2 in a humidified incubator. After 15 minutes, cells were either preincubated with anti–Plg-RKT mAb (280 nM; filled triangles, n = 6), isotype control antibody (280 nM; BD Biosciences; open squares, n = 3), or uPA aminoterminal fragment (50 ng/mL; Sekisui Diagnostics, Lexington, MA; filled diamonds, n = 4), or after an additional 30 minutes either EACA (0.2 M; open diamonds, n = 5) or aprotinin (2 µM; Sigma Aldrich; half-filled diamonds, n = 3) was added. Then plasminogen (0.4 µM; Roche) was added. Control cells were incubated for the times indicated above in vehicle only (n = 5). The number of migrated cells under this condition was set at 1, and values under experimental conditions are expressed as x-fold migration compared with control. As additional controls, cells were treated with anti–Plg-RKT mAb (280 nM; open triangles, n = 5) in the absence of plasminogen. Then, inserts were transferred into wells containing complete culture medium containing N-formyl-met-leu-phe (100 nM; Sigma Aldrich) as a chemoattractant. Cells were allowed to migrate for 3 hours at 37°C. Cells on the bottom side of the membrane were fixed with 4% paraformaldehyde and washed with phosphate-buffered saline. Membranes were then cut out and mounted on slides with Prolong Gold Antifade mounting medium (Thermo Fisher Scientific, Waltham, MA) and analyzed on a Zeiss Axio Imager fluorescence microscope. For calculating cell migration, cells on 5 randomly selected microscopic fields (magnification ×40) of the insert membrane were counted for every condition. Plasminogen-dependent migration was not exhibited by CMs or NCMs (CM: 0.9-fold vs control; NCM: 1.1-fold vs control). $P < .05 as compared with control; #not significant as compared with control; *P < .05, **P < .01, ***P < .001, as compared with plasminogen only; §not significant as compared with plasminogen only. (F) MFIs of uPAR on human monocyte subsets (n = 4). Cells were stained with anti-uPAR antibody (phycoerythrin; Clone VIM5; BioLegend, San Diego, CA). (G) Gating scheme for murine monocyte subsets in whole blood. Mouse monocytes were isolated from whole blood, and subsets were gated employing anti-CD11b and anti-Ly6C antibodies29 (Biolegend) using a Novocyte flow cytometer (ACEA Biosciences). (H) MFIs of Plg-RKT in murine monocyte subsets (n = 8; female, n = 4; male, n = 4). Whole blood samples were stained with anti-CD11b (allophycocyanin; Clone M1/70, BioLegend), anti-Ly6C (Phycoerythrin; Clone HK1.4, BioLegend), and anti–Plg-RKT antibody mAb.6,7 (I) Plasminogen binding to murine monocyte subsets in whole blood from female Plg-RKT+/+ wild-type (WT) mice and Plg-RKT−/− mice incubated with FITC-labeled plasminogen (0.5 µM; n = 3). Plg-RKT gene targeted mice (8-10 weeks of age) were backcrossed 10 generations into the C57Bl/6J background. (All animal experiments were approved by the Institutional Animal Care and Use Committee of The Scripps Research Institute.) (J) MFIs of Plg-RKT cell surface expression on murine male and female monocyte subsets. Whole blood samples were stained with anti-CD11b, anti-Ly6C, and anti–Plg-RKT as described in the legend to panel H (n = 8; 4 male and 4 female). (K) Ly6Chigh and (inset) Ly6Clow monocytes recovered in the peritoneal lavage of female Plg-RKT+/+ WT mice and Plg-RKT−/− mice either untreated (triangles) or 72 hours following intraperitoneal (IP) injection with thioglycollate (circles), as used previously in our laboratory7,8 (without thioglycollate treatment: n = 6 mice per group; with thioglycollate treatment: n = 5 mice per group). (L) Ly6Chigh and (inset) Ly6Clow monocytes recovered in the peritoneal lavage of male Plg-RKT+/+ WT mice and Plg-RKT−/− mice either untreated (triangles) or 72 hours following IP injection with thioglycollate (circles) (without thioglycollate treatment: n = 6 mice per group; with thioglycollate treatment: n = 5 mice per group). (M) Gating scheme for murine monocyte subsets in peritoneal lavage 72 hours following IP injection with thioglycollate. Mouse monocytes were isolated from peritoneal lavage and gated employing anti-CD11b and F4/80 antibodies (Biolegend) using a Novocyte flow cytometer (ACEA Biosciences). Values are displayed as MFIs (B,D,F, and H-J), delta cycle threshold (ΔCt) (C), fold change vs control (E), or total cell number (K,L) ± standard error of mean. Red symbols are used for CMs; blue symbols are used for IMs, and green symbols are used for NCMs for human monocytes (A-F). Blue symbols are used for Ly6Chigh, and red symbols are used for Ly6Clow murine monocytes (G-M). Statistical significances were calculated using analysis of variance and Tukey’s post hoc test when >2 groups were compared (B-F,I,K-L), and values of P < .05 were considered significant and are provided in the respective panels. Student t tests (unpaired) was used for comparison of 2 groups (H,J) and values of P < .05 were considered significant and are provided in the respective panels. FSC, forward scatter; n.s., not significant; SSC, sideward scatter.
Monocyte subsets and Plg-RKT. (A) Peripheral blood mononuclear cells were isolated from leukapheresis chambers obtained from healthy thrombocyte donors using lymphocyte separation medium 1077 (PromoCell, Heidelberg, Germany) and used for either monocyte subset isolation employing anti-CD45, anti-CD14, and anti-CD16 antibodies or for generating macrophages as described.15,26 Gating strategy for human monocyte subsets in whole blood is shown. The relative amounts of cells per subset were in accordance with published data, CM: 76% ± 5%, IM: 5% ± 2%, NCM: 19% ± 7%.15 (B) Mean fluorescent intensities (MFIs) of Plg-RKT on human monocyte subsets (n = 12). Monocytes in freshly drawn blood were stained with anti-CD14 antibody (PerCP/Cy5.5, Clone HCD14), anti-CD16 antibody (APC-Cy7, Clone 3G8), anti-CD45 antibody (Pacific Blue, Clone HI30), and anti–Plg-RKT mouse monoclonal mAb7H1, prepared and characterized in our laboratory.6,7 Samples were analyzed with a Novocyte Flow cytometer (ACEA Biosciences, San Diego, CA). (C) Plg-RKT mRNA levels in human monocyte subsets. Cells were sorted based on CD14 and CD16 expression into CMs, IM, and NCMs using a BD FACSAria (BD Biosciences, Franklin Lakes, NJ), and afterward, RNA was isolated for quantitative polymerase chain reaction. Primer sequences for Plg-RKT were “tggaacccttttagaaagaatga” and “ttggcagctgcaatttactc” (Universal Probe Library, #73; Roche, Basel, Switzerland). Glyceraldehyde-3-phosphate dehydrogenase was used for normalization (n = 3). (D) Plasminogen binding to human monocyte subsets in the presence or absence of anti–Plg-RKT mAb (clone7H1, developed and characterized in our laboratory)6,7 or EACA. PBMCs were stained with fluorochrome-labeled antibodies against CD45, CD14, and CD16 and incubated with FITC-labeled plasminogen (0.5 µM; n = 9).6 Fluorescence was measured with a Novocyte Flow cytometer. To test specificity of plasminogen binding, samples were additionally incubated with EACA (0.2 M; Sigma Aldrich, St. Louis, MO; n = 9) or with anti–Plg-RKT mAb (140 nM; n = 3). (E) Cell migration of human monocyte subsets in the presence or absence of plasminogen. Sorted IMs were seeded into inserts of Costar Transwell Permeable Supports (5.0-µm pore size; Corning, New York, NY) in RPMI 1640 containing 1% bovine serum albumin (both from Sigma Aldrich). Cells were incubated in total for 1 hour at 37°C, 5% CO2 in a humidified incubator. After 15 minutes, cells were either preincubated with anti–Plg-RKT mAb (280 nM; filled triangles, n = 6), isotype control antibody (280 nM; BD Biosciences; open squares, n = 3), or uPA aminoterminal fragment (50 ng/mL; Sekisui Diagnostics, Lexington, MA; filled diamonds, n = 4), or after an additional 30 minutes either EACA (0.2 M; open diamonds, n = 5) or aprotinin (2 µM; Sigma Aldrich; half-filled diamonds, n = 3) was added. Then plasminogen (0.4 µM; Roche) was added. Control cells were incubated for the times indicated above in vehicle only (n = 5). The number of migrated cells under this condition was set at 1, and values under experimental conditions are expressed as x-fold migration compared with control. As additional controls, cells were treated with anti–Plg-RKT mAb (280 nM; open triangles, n = 5) in the absence of plasminogen. Then, inserts were transferred into wells containing complete culture medium containing N-formyl-met-leu-phe (100 nM; Sigma Aldrich) as a chemoattractant. Cells were allowed to migrate for 3 hours at 37°C. Cells on the bottom side of the membrane were fixed with 4% paraformaldehyde and washed with phosphate-buffered saline. Membranes were then cut out and mounted on slides with Prolong Gold Antifade mounting medium (Thermo Fisher Scientific, Waltham, MA) and analyzed on a Zeiss Axio Imager fluorescence microscope. For calculating cell migration, cells on 5 randomly selected microscopic fields (magnification ×40) of the insert membrane were counted for every condition. Plasminogen-dependent migration was not exhibited by CMs or NCMs (CM: 0.9-fold vs control; NCM: 1.1-fold vs control). $P < .05 as compared with control; #not significant as compared with control; *P < .05, **P < .01, ***P < .001, as compared with plasminogen only; §not significant as compared with plasminogen only. (F) MFIs of uPAR on human monocyte subsets (n = 4). Cells were stained with anti-uPAR antibody (phycoerythrin; Clone VIM5; BioLegend, San Diego, CA). (G) Gating scheme for murine monocyte subsets in whole blood. Mouse monocytes were isolated from whole blood, and subsets were gated employing anti-CD11b and anti-Ly6C antibodies29 (Biolegend) using a Novocyte flow cytometer (ACEA Biosciences). (H) MFIs of Plg-RKT in murine monocyte subsets (n = 8; female, n = 4; male, n = 4). Whole blood samples were stained with anti-CD11b (allophycocyanin; Clone M1/70, BioLegend), anti-Ly6C (Phycoerythrin; Clone HK1.4, BioLegend), and anti–Plg-RKT antibody mAb.6,7 (I) Plasminogen binding to murine monocyte subsets in whole blood from female Plg-RKT+/+ wild-type (WT) mice and Plg-RKT−/− mice incubated with FITC-labeled plasminogen (0.5 µM; n = 3). Plg-RKT gene targeted mice (8-10 weeks of age) were backcrossed 10 generations into the C57Bl/6J background. (All animal experiments were approved by the Institutional Animal Care and Use Committee of The Scripps Research Institute.) (J) MFIs of Plg-RKT cell surface expression on murine male and female monocyte subsets. Whole blood samples were stained with anti-CD11b, anti-Ly6C, and anti–Plg-RKT as described in the legend to panel H (n = 8; 4 male and 4 female). (K) Ly6Chigh and (inset) Ly6Clow monocytes recovered in the peritoneal lavage of female Plg-RKT+/+ WT mice and Plg-RKT−/− mice either untreated (triangles) or 72 hours following intraperitoneal (IP) injection with thioglycollate (circles), as used previously in our laboratory7,8 (without thioglycollate treatment: n = 6 mice per group; with thioglycollate treatment: n = 5 mice per group). (L) Ly6Chigh and (inset) Ly6Clow monocytes recovered in the peritoneal lavage of male Plg-RKT+/+ WT mice and Plg-RKT−/− mice either untreated (triangles) or 72 hours following IP injection with thioglycollate (circles) (without thioglycollate treatment: n = 6 mice per group; with thioglycollate treatment: n = 5 mice per group). (M) Gating scheme for murine monocyte subsets in peritoneal lavage 72 hours following IP injection with thioglycollate. Mouse monocytes were isolated from peritoneal lavage and gated employing anti-CD11b and F4/80 antibodies (Biolegend) using a Novocyte flow cytometer (ACEA Biosciences). Values are displayed as MFIs (B,D,F, and H-J), delta cycle threshold (ΔCt) (C), fold change vs control (E), or total cell number (K,L) ± standard error of mean. Red symbols are used for CMs; blue symbols are used for IMs, and green symbols are used for NCMs for human monocytes (A-F). Blue symbols are used for Ly6Chigh, and red symbols are used for Ly6Clow murine monocytes (G-M). Statistical significances were calculated using analysis of variance and Tukey’s post hoc test when >2 groups were compared (B-F,I,K-L), and values of P < .05 were considered significant and are provided in the respective panels. Student t tests (unpaired) was used for comparison of 2 groups (H,J) and values of P < .05 were considered significant and are provided in the respective panels. FSC, forward scatter; n.s., not significant; SSC, sideward scatter.
In murine blood, the relative amounts of Ly6Chigh and of Ly6Clow cells were 32% ± 9% and 68% ± 9%, respectively (Figure 1G), which contrasts to some published data.18,19 However, according to a recent review, in mice the 2 populations each account for about half of the total monocytes and an increase in Ly6Clow cells mobilized from the marginal pool could be due to blood drawn under stress, such as cardiac puncture under terminal anesthesia, as used by us.20 In murine whole blood samples, the proinflammatory Ly6Chigh subset of monocytes21 expressed significantly higher levels of Plg-RKT than the Ly6Clow subset (Figure 1H). Correspondingly, the Plg-RKT+/+ Ly6Chigh subset bound significantly more plasminogen than the Plg-RKT+/+ Ly6Clow monocyte subset (Figure 1I). Furthermore, both subsets of Plg-RKT+/+ monocytes bound significantly more plasminogen than the respective Plg-RKT−/− monocyte subsets. Taken together, these results also suggest specific binding of plasminogen to Plg-RKT (Figure 1I). Notably, female murine monocytes expressed significantly higher levels of Plg-RKT than male murine monocytes (Figure 1J). Interestingly, circulating plasminogen levels are significantly higher in female compared with male mice8 and in female humans compared with male humans, suggesting the presence of a more efficient cell-associated plasminogen activation activity in female patients.22
To evaluate the in vivo relevance of our results, we used a sterile peritonitis model and determined the number of peritoneal cells in Plg-RKT+/+ and Plg-RKT−/− mice either untreated or 72 hours following thioglycollate injection.7,8 In untreated mice, no significant difference in the number of Ly6Chigh cells in the peritoneal lavage between Plg-RKT+/+ and Plg-RKT−/− mice was seen. When Plg-RKT+/+ mice were injected intraperitoneally with thioglycollate, a 24- and a 26-fold increase in Ly6Chigh cells in female and male Plg-RKT+/+ mice was observed, respectively, after thioglycollate injection compared with cell numbers without thioglycollate treatment. In contrast, this increase in numbers of Ly6Chigh cells after thioglycollate injection was more than halved to only 11- and 10-fold in Plg-RKT−/− female and male mice, respectively (ie, significantly less), namely, 46% and 57% less Ly6Chigh cells were present in the peritoneum of Plg-RKT−/− female and male mice, respectively, compared with the numbers of Ly6Chigh cells in Plg-RKT+/+ mice (Figure 1K-L). Notably, there were no effects of genotype on recruitment of Ly6Clow cells (Figure 1K-L insets), consistent with lower expression of Plg-RKT and lower plasminogen binding capacity of Ly6Clow cells (Figure 1H-I). Peritoneal IL-6 levels did not differ between untreated Plg-RKT+/+ and Plg-RKT−/− mice: 1.3 ± 0.1 pg/mg and 1.3 ± 0.1 pg/mg of peritoneal fluid protein for Plg-RKT+/+ and Plg-RKT−/− mice, respectively (P = n.s., n = 6 per group). IL-6 values were substantially increased in thioglycollate-treated animals, but the response was significantly attenuated in Plg-RKT−/− mice: 9.3 ± 1.7 pg/mg and 2.4 ± 0.8 pg/mg of peritoneal fluid protein for Plg-RKT+/+ and Plg-RKT−/− mice, respectively (P = .0019, n = 10 per group). Peritoneal IL-10 levels were below the detection limit of the assay used in untreated mice irrespective of genotype, but were significantly lower in thioglycollate-treated in Plg-RKT−/− mice as compared with Plg-RKT+/+ mice: 76.6 ± 11.6 pg/mg and 24.2 ± 6.7 pg/mg for thioglycollate-treated Plg-RKT+/+ and Plg-RKT−/− mice, respectively (P = .001, n = 10 per group).
The relative percentages of Ly6Chigh cells and of Ly6Clow cells positive for the macrophage marker, F4/80, were 89% and 92%, respectively (Figure 1M). Transcriptional profiling has shown that Ly6Clow monocytes extravasated into the peritoneum differentiate toward alternatively activated M2-like macrophages, whereas Ly6Chigh monocytes differentiate into inflammatory M1-like macrophages.23 This suggests that this differentiation process is a continuum from monocytes transitioning into the respective macrophage subsets and also suggests that the cells present in the peritoneal lavage following thioglycollate injection are in this transitioning process from monocytes to macrophages.
Monocytes are key effectors in the innate immune system and are differentiated to macrophages that are crucial for host defense and wound healing. Macrophages also play a key role in the pathogenesis of chronic inflammatory pathologies,24,25 where they are recruited to sites of inflammation and are capable of polarizing into functional subsets depending on environmental cues. We showed recently that matrix degradation depends on membrane-bound proteases and the expression of uPAR on proinflammatory macrophages and that alternatively activated macrophages are rendered proteolytically quiescent through their high expression of PAI-1.26 Therefore, we investigated whether differences in the expression of Plg-RKT might also exist in differentially polarized human macrophages and if such differential expression might impact the migratory capacity of these cells.
Proinflammatory macrophages expressed significantly more cell surface Plg-RKT and Plg-RKT messenger RNA (mRNA) compared with alternatively activated macrophages or unpolarized macrophages (Figure 2A-B).
![Figure 2. Proinflammatory macrophages express high levels of Plg-RKT in vitro and in vivo. (A) MFIs of Plg-RKT on human unpolarized macrophages (M0), human macrophages polarized with LPS (Sigma Aldrich) and IFN-γ (Thermo Fisher Scientific) [M(LPS+IFN)] and human macrophages polarized with IL-4 (Thermo Fisher Scientific) and IL-13 (Santa Cruz, Dallas, TX) [M(IL-4+IL-13)] (n = 5). Unpolarized and polarized macrophages were stained with anti–Plg-RKT mAb and analyzed on a Novocyte flow cytometer (ACEA Biosciences). (B) Plg-RKT mRNA levels in human unpolarized macrophages (M0), human macrophages polarized with LPS and IFN-γ [M(LPS+IFN)], and human macrophages polarized with IL-4 and IL-13 [M(IL-4+IL-13)] (n = 3). (C) Unpolarized and polarized macrophages were seeded into Costar Transwell Permeable Supports (5.0-µm pore size; Corning) in complete culture medium consisting of RPMI 1640 containing 20% fetal bovine serum (Biochrome Millipore, Berlin, Germany). Cells were incubated for 1 hour at 37°C, 5% CO2 in a humidified incubator. Cells were either preincubated for 30 minutes with anti–Plg-RKT mAb (140 nm) or with vehicle. Thereafter, plasminogen (0.2 µM; Roche) or vehicle was added or cells were left without the addition of either mAb or plasminogen as control. Then, the medium was removed, replaced by phosphate-buffered saline, pH 7.4, containing the respective additions or no additions, and inserts were transferred into wells containing complete culture medium. Cells were allowed to migrate for 3 hours at 37°C. Quantification of migrated cells was performed as described for monocytes in the legend to panel E of Figure 1 (n = 5). (D-E) Representative images of immunohistochemical staining of human carotid artery sections (D; n = 15) and adipose tissue sections (E; n = 8) stained for CD80, Plg-RKT, and plasminogen. Carotid plaque samples were obtained as described.30 Visceral fat tissue was obtained from 8 patients undergoing bariatric bypass surgery.31 Tissue samples were fixed in 4% formaldehyde and embedded in paraffin. Sections of human carotid artery plaques were stained with hematoxylin and eosin, and sections of human carotid artery plaques and of adipose tissue samples were stained with an anti-CD80 antibody as a marker for M(LPS+INF) macrophages (rabbit polyclonal, dilution 1:100; Santa Cruz), anti–Plg-RKT mAB (dilution 1:500), an antiplasminogen antibody (goat polyclonal; dilution 1:100; Santa Cruz), and a respective isotype control antibody for the anti–Plg-RKT mAb (dilution 1:250; eBiosciences, San Diego, CA) and normal goat IgG as control antibody for the antiplasminogen antibody (dilution 1:750; R&D Systems, Minneapolis, MN). Afterward, sections were incubated for 18 hours with secondary antibodies (anti-rabbit polyclonal, labeled with Cy3; Biolegend; anti-goat polyclonal, labeled with Dylight 650 and anti-mouse polyclonal labeled with FITC, dilution 1:200; both Abcam). Slides were scanned on a fully automated system (TissueGnostics, Vienna, Austria) with a ×20 objective, acquired with tissueFAX software and analyzed with tissueQuest software. CD80 is depicted in red; Plg-RKT is depicted in green, and plasminogen is depicted in yellow. White arrows in the merged panel mark cells staining positive for CD80, Plg-RKT, and Plg. Cells with MFI >100 for CD80 were defined as CD80 high-expressing cells and therefore as proinflammatory; cells with MFI <100 were defined as anti-inflammatory. MFIs for Plg-RKT and plasminogen were then measured for the 2 defined CD80 high- and low-expressing cells, respectively, and compared. Values are displayed as MFIs (A), ΔCt (B), or fold change vs control (C) ± standard error of mean. Statistical significances were calculated using analysis of variance and Tukey’s post hoc test when >2 groups were compared (A-C) or Student t tests (unpaired) for comparison of 2 groups (D,E). DAPI, 4′,6-diamidino-2-phenylindole.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/134/6/10.1182_blood.2018850420/3/m_blood850420f2.png?Expires=1769802503&Signature=lYq-hcs0iym6D2FeM2yRr~GjgTsJcFB1YuygZJIOci-cLnsA7gPGnvxjAE5Xq-B4AeUsQhzzwMr-QfgpRMkyCCrZs-hPuX7zjYhCpe6xF5-D3n6sN~oOT6mZhpGvDFKcA3xbLTShAbGVmL--CH-JkkLVYgi4gvrKK3uyuiy1ucMgvb6jN3hZnXzKH-eksOd3BUey4uX0ZaMls1oUpBZcR~Qk~huqGRlDN952SvAUdR6WHwpsoQ5tUwby7uLJGIrcfX11Mw7zpxArhyVOM9CYCpQv8YPsGWCcul6A4DpjUa0oq6RUwSsh7cUni8y1~fwmupWKh74g8Ed1dhh52YP72g__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Proinflammatory macrophages express high levels of Plg-RKT in vitro and in vivo. (A) MFIs of Plg-RKT on human unpolarized macrophages (M0), human macrophages polarized with LPS (Sigma Aldrich) and IFN-γ (Thermo Fisher Scientific) [M(LPS+IFN)] and human macrophages polarized with IL-4 (Thermo Fisher Scientific) and IL-13 (Santa Cruz, Dallas, TX) [M(IL-4+IL-13)] (n = 5). Unpolarized and polarized macrophages were stained with anti–Plg-RKT mAb and analyzed on a Novocyte flow cytometer (ACEA Biosciences). (B) Plg-RKT mRNA levels in human unpolarized macrophages (M0), human macrophages polarized with LPS and IFN-γ [M(LPS+IFN)], and human macrophages polarized with IL-4 and IL-13 [M(IL-4+IL-13)] (n = 3). (C) Unpolarized and polarized macrophages were seeded into Costar Transwell Permeable Supports (5.0-µm pore size; Corning) in complete culture medium consisting of RPMI 1640 containing 20% fetal bovine serum (Biochrome Millipore, Berlin, Germany). Cells were incubated for 1 hour at 37°C, 5% CO2 in a humidified incubator. Cells were either preincubated for 30 minutes with anti–Plg-RKT mAb (140 nm) or with vehicle. Thereafter, plasminogen (0.2 µM; Roche) or vehicle was added or cells were left without the addition of either mAb or plasminogen as control. Then, the medium was removed, replaced by phosphate-buffered saline, pH 7.4, containing the respective additions or no additions, and inserts were transferred into wells containing complete culture medium. Cells were allowed to migrate for 3 hours at 37°C. Quantification of migrated cells was performed as described for monocytes in the legend to panel E of Figure 1 (n = 5). (D-E) Representative images of immunohistochemical staining of human carotid artery sections (D; n = 15) and adipose tissue sections (E; n = 8) stained for CD80, Plg-RKT, and plasminogen. Carotid plaque samples were obtained as described.30 Visceral fat tissue was obtained from 8 patients undergoing bariatric bypass surgery.31 Tissue samples were fixed in 4% formaldehyde and embedded in paraffin. Sections of human carotid artery plaques were stained with hematoxylin and eosin, and sections of human carotid artery plaques and of adipose tissue samples were stained with an anti-CD80 antibody as a marker for M(LPS+INF) macrophages (rabbit polyclonal, dilution 1:100; Santa Cruz), anti–Plg-RKT mAB (dilution 1:500), an antiplasminogen antibody (goat polyclonal; dilution 1:100; Santa Cruz), and a respective isotype control antibody for the anti–Plg-RKT mAb (dilution 1:250; eBiosciences, San Diego, CA) and normal goat IgG as control antibody for the antiplasminogen antibody (dilution 1:750; R&D Systems, Minneapolis, MN). Afterward, sections were incubated for 18 hours with secondary antibodies (anti-rabbit polyclonal, labeled with Cy3; Biolegend; anti-goat polyclonal, labeled with Dylight 650 and anti-mouse polyclonal labeled with FITC, dilution 1:200; both Abcam). Slides were scanned on a fully automated system (TissueGnostics, Vienna, Austria) with a ×20 objective, acquired with tissueFAX software and analyzed with tissueQuest software. CD80 is depicted in red; Plg-RKT is depicted in green, and plasminogen is depicted in yellow. White arrows in the merged panel mark cells staining positive for CD80, Plg-RKT, and Plg. Cells with MFI >100 for CD80 were defined as CD80 high-expressing cells and therefore as proinflammatory; cells with MFI <100 were defined as anti-inflammatory. MFIs for Plg-RKT and plasminogen were then measured for the 2 defined CD80 high- and low-expressing cells, respectively, and compared. Values are displayed as MFIs (A), ΔCt (B), or fold change vs control (C) ± standard error of mean. Statistical significances were calculated using analysis of variance and Tukey’s post hoc test when >2 groups were compared (A-C) or Student t tests (unpaired) for comparison of 2 groups (D,E). DAPI, 4′,6-diamidino-2-phenylindole.
Proinflammatory macrophages express high levels of Plg-RKT in vitro and in vivo. (A) MFIs of Plg-RKT on human unpolarized macrophages (M0), human macrophages polarized with LPS (Sigma Aldrich) and IFN-γ (Thermo Fisher Scientific) [M(LPS+IFN)] and human macrophages polarized with IL-4 (Thermo Fisher Scientific) and IL-13 (Santa Cruz, Dallas, TX) [M(IL-4+IL-13)] (n = 5). Unpolarized and polarized macrophages were stained with anti–Plg-RKT mAb and analyzed on a Novocyte flow cytometer (ACEA Biosciences). (B) Plg-RKT mRNA levels in human unpolarized macrophages (M0), human macrophages polarized with LPS and IFN-γ [M(LPS+IFN)], and human macrophages polarized with IL-4 and IL-13 [M(IL-4+IL-13)] (n = 3). (C) Unpolarized and polarized macrophages were seeded into Costar Transwell Permeable Supports (5.0-µm pore size; Corning) in complete culture medium consisting of RPMI 1640 containing 20% fetal bovine serum (Biochrome Millipore, Berlin, Germany). Cells were incubated for 1 hour at 37°C, 5% CO2 in a humidified incubator. Cells were either preincubated for 30 minutes with anti–Plg-RKT mAb (140 nm) or with vehicle. Thereafter, plasminogen (0.2 µM; Roche) or vehicle was added or cells were left without the addition of either mAb or plasminogen as control. Then, the medium was removed, replaced by phosphate-buffered saline, pH 7.4, containing the respective additions or no additions, and inserts were transferred into wells containing complete culture medium. Cells were allowed to migrate for 3 hours at 37°C. Quantification of migrated cells was performed as described for monocytes in the legend to panel E of Figure 1 (n = 5). (D-E) Representative images of immunohistochemical staining of human carotid artery sections (D; n = 15) and adipose tissue sections (E; n = 8) stained for CD80, Plg-RKT, and plasminogen. Carotid plaque samples were obtained as described.30 Visceral fat tissue was obtained from 8 patients undergoing bariatric bypass surgery.31 Tissue samples were fixed in 4% formaldehyde and embedded in paraffin. Sections of human carotid artery plaques were stained with hematoxylin and eosin, and sections of human carotid artery plaques and of adipose tissue samples were stained with an anti-CD80 antibody as a marker for M(LPS+INF) macrophages (rabbit polyclonal, dilution 1:100; Santa Cruz), anti–Plg-RKT mAB (dilution 1:500), an antiplasminogen antibody (goat polyclonal; dilution 1:100; Santa Cruz), and a respective isotype control antibody for the anti–Plg-RKT mAb (dilution 1:250; eBiosciences, San Diego, CA) and normal goat IgG as control antibody for the antiplasminogen antibody (dilution 1:750; R&D Systems, Minneapolis, MN). Afterward, sections were incubated for 18 hours with secondary antibodies (anti-rabbit polyclonal, labeled with Cy3; Biolegend; anti-goat polyclonal, labeled with Dylight 650 and anti-mouse polyclonal labeled with FITC, dilution 1:200; both Abcam). Slides were scanned on a fully automated system (TissueGnostics, Vienna, Austria) with a ×20 objective, acquired with tissueFAX software and analyzed with tissueQuest software. CD80 is depicted in red; Plg-RKT is depicted in green, and plasminogen is depicted in yellow. White arrows in the merged panel mark cells staining positive for CD80, Plg-RKT, and Plg. Cells with MFI >100 for CD80 were defined as CD80 high-expressing cells and therefore as proinflammatory; cells with MFI <100 were defined as anti-inflammatory. MFIs for Plg-RKT and plasminogen were then measured for the 2 defined CD80 high- and low-expressing cells, respectively, and compared. Values are displayed as MFIs (A), ΔCt (B), or fold change vs control (C) ± standard error of mean. Statistical significances were calculated using analysis of variance and Tukey’s post hoc test when >2 groups were compared (A-C) or Student t tests (unpaired) for comparison of 2 groups (D,E). DAPI, 4′,6-diamidino-2-phenylindole.
Furthermore, proinflammatory macrophages exhibited plasminogen-dependent cell migration that was impaired by treatment with anti–Plg-RKT mAb, whereas significant plasminogen-dependent migration was not exhibited by alternatively activated or unpolarized macrophages (Figure 2C).
To further evaluate the relevance of our in vitro findings, under in vivo inflammatory conditions, sections of human carotid artery plaques obtained from patients undergoing atherectomy and sections of adipose tissue obtained from morbidly obese patients undergoing gastric bypass surgery were stained with antibodies against the inflammatory macrophage marker CD80, Plg-RKT, and plasminogen. CD80high proinflammatory macrophages expressed significantly more Plg-RKT that was colocalized with plasminogen compared with CD80low macrophages in human carotid artery plaques and obese adipose tissue (Figure 2D-E). When these sections were stained with the markers for alternatively activated macrophages CD206 or CD209, no association between these markers and Plg-RKT expression was found (data not shown).
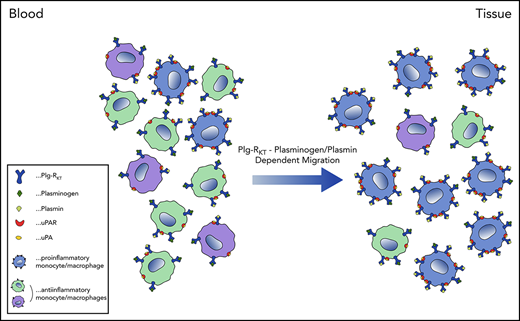
We here provide the first evidence that Plg-RKT is differentially expressed on monocyte and macrophage subsets, with inflammatory IMs and proinflammatory macrophages expressing the highest levels of this plasminogen receptor. Differential expression of Plg-RKT impacted the plasminogen- and plasmin-dependent migratory capacity of these cells. Proinflammatory macrophages expressed significantly more Plg-RKT in human atherosclerotic plaques and obese adipose tissue than macrophages expressing low levels of the inflammatory marker CD80. Finally, recruitment of Ly6Chigh monocytes was impaired in Plg-RKT–deficient mice, concomitantly with impairment in levels of the representative cytokines, the proinflammatory cytokine IL-6, and IL-10, which has anti-inflammatory properties, but is induced frequently in inflammatory situations. 27,28 Our results suggest that higher expression of Plg-RKT on proinflammatory monocyte and macrophage subsets contributes significantly to their migration and recruitment into areas of inflammation and their regulation of the inflammatory response in pathologies characterized by an inflammatory state.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank the Core Facilities for Flow Cytometry at Scripps Research Institute, La Jolla, CA, and the Core Facilities of the Medical University of Vienna, Vienna, Austria for their expertise in cell sorting.
This project was funded by the Austrian Science Fund (FWF; SFB-54) (J.W. and W.S.S.), by National Institutes of Health, National Heart, Lung, and Blood Institute grant HL 081046 (L.A.M.), and by Merit Review Award 5I01BX003933 from the US Department of Veterans Affairs (R.J.P.). B.T. received a Travel Grant from the International Society of Fibrinolysis and Proteolysis.
Authorship
Contribution: B.T. performed the majority of the experiments; J.B. and N.B. assisted in cell culture experiments; A.P. scanned the tissue slides and helped with the analysis; G.R.-K., C.K., and M.P. provided and characterized the adipose tissue samples; S.S., S.D., and I.H. provided and characterized the carotid artery plaques; M.B.F. provided the leukapheresis chambers and critically revised the manuscript; the experiments were designed and analyzed by B.T., P.J.H., W.S.S., L.A.M., R.J.P., and J.W.; and the manuscript was written by B.T. and critically revised and modified by P.J.H., K.H., R.J.P., W.S.S., J.W., and L.A.M.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Lindsey A. Miles, Department of Molecular Medicine, The Scripps Research Institute, 10550 N. Torrey Pines Rd, SP30-3020, La Jolla, CA 92037; e-mail: lmiles@scripps.edu.

![Figure 2. Proinflammatory macrophages express high levels of Plg-RKT in vitro and in vivo. (A) MFIs of Plg-RKT on human unpolarized macrophages (M0), human macrophages polarized with LPS (Sigma Aldrich) and IFN-γ (Thermo Fisher Scientific) [M(LPS+IFN)] and human macrophages polarized with IL-4 (Thermo Fisher Scientific) and IL-13 (Santa Cruz, Dallas, TX) [M(IL-4+IL-13)] (n = 5). Unpolarized and polarized macrophages were stained with anti–Plg-RKT mAb and analyzed on a Novocyte flow cytometer (ACEA Biosciences). (B) Plg-RKT mRNA levels in human unpolarized macrophages (M0), human macrophages polarized with LPS and IFN-γ [M(LPS+IFN)], and human macrophages polarized with IL-4 and IL-13 [M(IL-4+IL-13)] (n = 3). (C) Unpolarized and polarized macrophages were seeded into Costar Transwell Permeable Supports (5.0-µm pore size; Corning) in complete culture medium consisting of RPMI 1640 containing 20% fetal bovine serum (Biochrome Millipore, Berlin, Germany). Cells were incubated for 1 hour at 37°C, 5% CO2 in a humidified incubator. Cells were either preincubated for 30 minutes with anti–Plg-RKT mAb (140 nm) or with vehicle. Thereafter, plasminogen (0.2 µM; Roche) or vehicle was added or cells were left without the addition of either mAb or plasminogen as control. Then, the medium was removed, replaced by phosphate-buffered saline, pH 7.4, containing the respective additions or no additions, and inserts were transferred into wells containing complete culture medium. Cells were allowed to migrate for 3 hours at 37°C. Quantification of migrated cells was performed as described for monocytes in the legend to panel E of Figure 1 (n = 5). (D-E) Representative images of immunohistochemical staining of human carotid artery sections (D; n = 15) and adipose tissue sections (E; n = 8) stained for CD80, Plg-RKT, and plasminogen. Carotid plaque samples were obtained as described.30 Visceral fat tissue was obtained from 8 patients undergoing bariatric bypass surgery.31 Tissue samples were fixed in 4% formaldehyde and embedded in paraffin. Sections of human carotid artery plaques were stained with hematoxylin and eosin, and sections of human carotid artery plaques and of adipose tissue samples were stained with an anti-CD80 antibody as a marker for M(LPS+INF) macrophages (rabbit polyclonal, dilution 1:100; Santa Cruz), anti–Plg-RKT mAB (dilution 1:500), an antiplasminogen antibody (goat polyclonal; dilution 1:100; Santa Cruz), and a respective isotype control antibody for the anti–Plg-RKT mAb (dilution 1:250; eBiosciences, San Diego, CA) and normal goat IgG as control antibody for the antiplasminogen antibody (dilution 1:750; R&D Systems, Minneapolis, MN). Afterward, sections were incubated for 18 hours with secondary antibodies (anti-rabbit polyclonal, labeled with Cy3; Biolegend; anti-goat polyclonal, labeled with Dylight 650 and anti-mouse polyclonal labeled with FITC, dilution 1:200; both Abcam). Slides were scanned on a fully automated system (TissueGnostics, Vienna, Austria) with a ×20 objective, acquired with tissueFAX software and analyzed with tissueQuest software. CD80 is depicted in red; Plg-RKT is depicted in green, and plasminogen is depicted in yellow. White arrows in the merged panel mark cells staining positive for CD80, Plg-RKT, and Plg. Cells with MFI >100 for CD80 were defined as CD80 high-expressing cells and therefore as proinflammatory; cells with MFI <100 were defined as anti-inflammatory. MFIs for Plg-RKT and plasminogen were then measured for the 2 defined CD80 high- and low-expressing cells, respectively, and compared. Values are displayed as MFIs (A), ΔCt (B), or fold change vs control (C) ± standard error of mean. Statistical significances were calculated using analysis of variance and Tukey’s post hoc test when >2 groups were compared (A-C) or Student t tests (unpaired) for comparison of 2 groups (D,E). DAPI, 4′,6-diamidino-2-phenylindole.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/134/6/10.1182_blood.2018850420/3/m_blood850420f2.png?Expires=1769802504&Signature=DtdBjJlm3CKQwxoq4ZMq4S7Yi8LKNsET1C3dqzu99UaWrot9y4~FebgbA68LzzpJVuVK3-lUqQGuWxDtvhqLGdKHvTj7wRyp5cgbNbgxfR7pdWsxiwDCxRD7im4QQeONjjhthI6gYLXiv7Ls1A8cBlC0u2sgz1XB7MuNCXV9n941sbAMBi4e0GELhh-kN-yKhCI7kE2TDgSknS9eFcnyDAWNJw~VRcakEWSy9pAn5ksKliHTveZSgOMzlG84nwnkCKZGx5227SmdIamlxRfMYLqg~WxdvtkDGJfj3gBbTaj1r9TRG-2ZKDfpQozhuaqUXKoBOrQ5dVljlmgXA8J3-g__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
