

In this issue of Blood, 1 provide molecular insights into how dynamic changes in cohesin complex levels are critical to erythroid differentiation and how perturbations can contribute to myeloid malignancies.
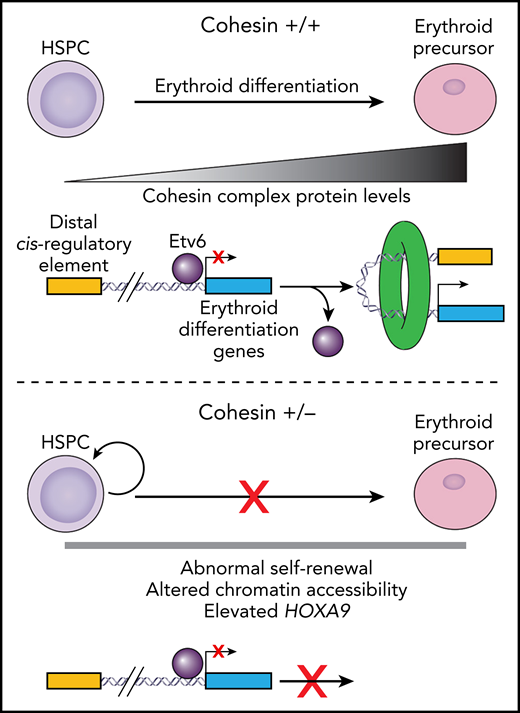
In cohesin wild-type (+/+) cells, HSPC differentiation down the erythroid lineage requires a gradual increase in cohesin protein levels (top). This increase in cohesin evicts the transcriptional repressor Etv6 (purple circle) from genes required for erythroid differentiation (blue box). In addition, it facilitates the formation of chromatin loops permitting distal cis-regulatory elements (yellow box) and erythroid differentiation genes to interact, resulting in gene activation. In cohesin haploinsufficient cells (+/−), cohesin levels cannot increase during erythroid commitment (bottom). This prevents the eviction of Etv6 and induction of genes required for erythroid differentiation.
In cohesin wild-type (+/+) cells, HSPC differentiation down the erythroid lineage requires a gradual increase in cohesin protein levels (top). This increase in cohesin evicts the transcriptional repressor Etv6 (purple circle) from genes required for erythroid differentiation (blue box). In addition, it facilitates the formation of chromatin loops permitting distal cis-regulatory elements (yellow box) and erythroid differentiation genes to interact, resulting in gene activation. In cohesin haploinsufficient cells (+/−), cohesin levels cannot increase during erythroid commitment (bottom). This prevents the eviction of Etv6 and induction of genes required for erythroid differentiation.
Hematologic cancers derived from myeloid cells, such as myelodysplastic syndromes (MDS) or acute myeloid leukemia, remain high-risk diseases with suboptimal long-term survival. Importantly, in myeloid malignancies, mutations within genes encoding the cohesin complex (RAD21, SMC3, SMC1A, and STAG2) occur within ∼15% of patients (for a review, see Fisher et al2 ), making them comparable in frequency to mutations in other “drivers” such as KRAS/NRAS or TET2.3 The cohesin complex has a well-described role in mitosis, where it promotes sister chromatid cohesion and is expressed in all cell types, including postmitotic cells such as neurons. Recently, it has been discovered that the cohesin complex also plays a critical role in the regulation of gene expression by facilitating the interaction of distal DNA regulatory sequences with genes by “looping out” the intervening chromatin segment.4 Complete loss of the cohesin complex prevents proper mitosis and quickly induces cell death, so not surprisingly, almost all mutations of cohesin genes in myeloid malignancies are heterozygous, or in the case of STAG2, complete loss of function is partially tolerated because of compensation by its paralog, STAG1, as elegantly demonstrated in a recent report.5 Multiple groups6-9 have reported the phenotype induced by cohesin haploinsufficiency with remarkable agreement that loss of cohesin enhances hematopoietic stem and progenitor cell (HSPC) self-renewal, a critical first step in the development myeloid malignancies. These studies also identified that altered chromatin accessibility6-8 and/or elevated expression of the transcription factor HOXA99 were key drivers of this abnormal HSPC self-renewal. Importantly, all the proposed mechanisms of augmenting self-renewal in HSPCs assume that cohesin levels are relatively invariant across cell types, implying that heterozygous mutations should affect different hematopoietic lineages similarly, although this is clearly not the case and has been an important open question within the field.
The elegant study performed by Sasca et al in this issue of Blood challenges the concept that all hematopoietic lineages express cohesin proteins at the same level. Using a combination of genomics, single-cell transcriptomics, mass spectrometry–based proteomics, and patient-derived data, they identified not only a novel phenotype associated with cohesin loss but also the underlying molecular mechanism. This study demonstrates that cohesin protein levels rise as HSPCs differentiate into the erythroid lineage (see figure). In HSPCs, cohesin binds to genes required for erythroid differentiation occupied by the transcriptional repressor Etv6, which prevents their expression. Upon addition of erythropoietin to stimulate differentiation of HSPCs, cohesin accumulates at these same Etv6-occupied red cell genes, and as cohesin levels rise, Etv6 is displaced from chromatin, thereby permitting these genes to be activated and thereby initiate erythropoiesis. The precise mechanism by which Etv6 is displaced from chromatin is unknown and needs to be clarified through future studies. In the case of heterozygous cohesin mutations, the loss of an allele prevents the dynamic increase in cohesin protein levels required for erythropoiesis. To verify that a similar phenotype is observed in patients with myeloid malignancies, the authors queried published data sets on primary samples and observed an impairment in erythroid-specific gene expression programs in patients with cohesin mutations compared with other patients. Collectively, the authors reveal how dynamic changes in cohesin levels are critical to cell-fate decisions by facilitating alterations in gene expression. Importantly, the mechanism that promotes altered cohesin levels is likely at the level of gene expression, although a posttranscriptional mechanism cannot be ruled out.
While the authors did not evaluate HSPC self-renewal directly, one could speculate that a block in differentiation down the erythroid lineage could induce a homeostatic mechanism that ultimately leads to increased HSPC self-renewal. The augmented HSPC self-renewal proposed by previous studies6-9 and the erythroid-differentiation block described by Sasca et al are not mutually exclusive, and both mechanisms may be relevant to the clinical presentation of myeloid malignancies with cohesin mutations. In addition, the concept that cohesin levels vary across hematopoietic cell types has the potential to open new mechanisms by which cohesin affects the lineage commitment of HSPCs if it can be demonstrated in subsequent studies that different subsets of stem and progenitor cells also express varying levels of cohesin.
The molecular insights provided within this study are only possible through the leveraging of multiomic approaches by the group to address the details of how cohesin loss promotes myeloid malignancies. This required the use of a cell line to provide homogeneous subsets for the wealth of detailed molecular studies performed that ultimately revealed a novel molecular mechanism by which cohesin regulates erythroid differentiation. While Sasca et al used patient-derived data to verify key aspects of their findings, it will be important for further validation in primary human HSPCs and myeloid malignancy samples in conjunction with animal models to fully recapitulate hematopoiesis, both normal and malignant, in vivo. Nonetheless, by identifying a specific defect in erythroid lineage commitment as a potential consequence of cohesin mutations, one can speculate at potential therapeutic strategies in this subtype of myeloid malignancy patients. For example, therapies that promote differentiation down the erythroid lineage may ameliorate some aspects of the bone marrow defects seen in patients with myeloid malignancy, especially MDS. In addition, it may be therapeutically useful for reducing disease burden, as has been seen when differentiation agents are used without conventional chemotherapy in patients with acute promyelocytic leukemia.10
Conflict-of-interest disclosure: The authors declare no competing financial interests.

