Key Points
PDK1 is critically required for MK F-actin cytoskeleton dynamics during MK maturation, podosome formation, and DMS polarization.
PDK1-dependent signaling in MKs during proplatelet formation involves the PAK/LIM domain kinase /cofilin phosphorylation cascade and atypical PKC isoform ζ.

Abstract
During thrombopoiesis, megakaryocytes (MKs) form proplatelets within the bone marrow (BM) and release platelets into BM sinusoids. Phosphoinositide-dependent protein kinase-1 (PDK1) is required for Ca2+-dependent platelet activation, but its role in MK development and regulation of platelet production remained elusive. The present study explored the role of PDK1 in the regulation of MK maturation and polarization during thrombopoiesis using a MK/platelet-specific knockout approach. Pdk1-deficient mice (Pdk1−/−) developed a significant macrothrombocytopenia as compared with wild-type mice (Pdk1fl/fl). Pdk1 deficiency further dramatically increased the number of MKs without sinusoidal contact within the BM hematopoietic compartment, resulting in a pronounced MK hyperplasia and a significantly increased extramedullary thrombopoiesis. Cultured Pdk1−/− BM-MKs showed impaired spreading on collagen, associated with an altered actin cytoskeleton structure with less filamentous actin (F-actin) and diminished podosome formation, whereas the tubulin cytoskeleton remained unaffected. This phenotype was associated with abrogated phosphorylation of p21-activated kinase (PAK) as well as its substrates LIM domain kinase and cofilin, supporting the hypothesis that the defective F-actin assembly results from increased cofilin activity in Pdk1-deficient MKs. Pdk1−/− BM-MKs developed increased ploidy and exhibited an abnormal ultrastructure with disrupted demarcation membrane system (DMS). Strikingly, Pdk1−/− BM-MKs displayed a pronounced defect in DMS polarization and produced significantly less proplatelets, indicating that PDK1 is critically required for proplatelet formation. In human MKs, genetic PDK1 knockdown resulted in increased maturity but reduced platelet-like particles formation. The present observations reveal a pivotal role of PDK1 in the regulation of MK cytoskeletal dynamics and polarization, proplatelet formation, and thrombopoiesis.
Introduction
Platelets are critically involved in the pathogenesis of arterial thrombosis and essential for maintaining hemostasis and vascular integrity.1,2 These small anucleated cells derive from proplatelets, which are extended into sinusoids of the bone marrow (BM) by polyploid megakaryocytes (MKs).3 MKs develop from pluripotent hematopoietic stem cells and become polyploid during their maturation through repeated cycles of DNA replication without cell division, a process called endomitosis.4 During completion of endomitosis, MKs develop the demarcation membrane system (DMS), an endomembrane compartment that serves as a membrane resource for proplatelet production.5,6 Thereby, a distinct reorganization of the MK cytoskeleton and particularly the filamentous actin (F-actin) network is critical for DMS maturation and polarization of MKs toward BM sinusoids.7-10 MKs adjacent to BM sinusoids extend and release long branching cytoplasmatic protrusions (proplatelets) into the sinusoidal lumen, from which platelets are shed by the shear forces of the bloodstream.11,12 Defects in thrombopoiesis associated with impaired platelet production may result in abrogated hemostasis and severe bleeding disorders. However, although thrombocytopenia reflects a major clinical problem, the molecular mechanisms underlying MK cytoskeletal reorganization in megakaryopoiesis and platelet biogenesis remained elusive.
In the present study, we aimed to determine the role of phosphoinositide-dependent protein kinase 1 (PDK1) in MK cytoskeleton reorganization during MK maturation and platelet biogenesis.
PDK1 is a cytoplasmatic membrane-associated AGC serine/threonine kinase activated downstream of phosphoinositide 3-kinase (PI3K) that regulates several further AGC kinases, including protein kinase B/AKT and serum- and glucocorticoid-inducible kinase-1 as well as protein kinase A and protein kinase C (PKC).13-15 PDK1/AGC kinase signaling pathways regulate diverse cellular processes, such as those relevant to metabolism, development, proliferation, and survival.16,17 Genetic deletion of Pdk1 in mice causes embryonic lethality.16,18 PDK1 is an important regulator of actin cytoskeleton dynamics in dendritic cells, endothelial cell chemotaxis, and migration of cancer cells.14,19-21
In platelets, PDK1 mediates thrombin-induced platelet aggregation and clot retraction by regulating integrin αIIbβ3 outside-in signaling via Akt-dependent inhibition of glycogen synthase kinase-3β.22 PDK1 further determines collagen-triggered increase of intracellular Ca2+ and consecutive arterial thrombus formation via activation of phospholipase Cγ2.23 Moreover, PDK1 plays an important role in the cross talk between PI3K and mitogen-activated protein kinase signaling in adenosine diphosphate–induced platelet activation, thromboxane generation, and thrombosis.24 The cytokine thrombopoietin (TPO) promotes MK maturation via stimulation of the PI3K as well as mitogen-activated protein kinase signaling pathways.25,26 Recently, it has been reported that PDK1 may play a positive role in reactive thrombocytosis due to increased proplatelet formation following stimulation of MKs with the inflammatory chemokine RANTES.27 However, the role of PDK1 in megakaryopoiesis and platelet production has not been investigated so far.
In the present study, we aimed to determine the role of PDK1 in MK maturation, cytoskeleton reorganization, and polarization and its impact on proplatelet formation during thrombopoiesis in mice lacking PDK1 in the MK lineage.
Materials and methods
Mice
PDK1-floxed mice (Pdk1fl/fl) were generated by Dario Alessi (University of Dundee, UK). Origin, breeding, and genotyping of Pdk1fl/fl mice were described previously.16 For MK-specific deletion of Pdk1, Pdk1fl/fl mice were crossed with transgenic mice expressing Cre-recombinase under the control of the MK- and platelet-specific platelet factor 4 (Pf4-Cre) to obtain Pdk1fl/flpf4-Cre+ (Pdk1−/−) and Pdk1fl/flpf4-Cre− (Pdk1fl/fl) littermates as described previously.23 Mice were studied at the age of 8 to 12 weeks. All animal experiments were performed according to the German animal protection law and approved by the local authorities (Regierungspräsidium Tübingen).
Methods
In vitro differentiation and cultivation of fetal liver– as well as BM-derived MKs, real-time polymerase chain reaction, western blot analysis, determination of TPO levels, analysis of thrombopoiesis after TPO treatment, determination of platelet lifespan assay and recovery following platelet depletion, immunohistochemistry of murine BM and spleen, immunofluorescence staining of femora cryosections, confocal microscopy of MK polarization, ultrastructural transmission electron microscopy (TEM) analysis of MKs, immunofluorescence staining of cultured MKs, LifeAct imaging, determination of MK ploidy, in vitro immunostaining of proplatelet-forming BM MKs, culture, differentiation and analysis of human CD34+ cells, multiphoton intravital microscopy analysis of in vivo proplatelet formation, determination of RhoA and Cdc42 activity, determination of Caspase 3/7 activity, determination of hematopoietic progenitor cells, and statistical analysis are described in detail in the supplemental Material and methods, available on the Blood Web site.
Results
Genetic deletion of Pdk1 results in embryonic lethality in mice. Therefore, to specifically delete Pdk1 in the MK lineage and to circumvent prenatal death, Pdk1fl/fl mice were crossed with transgenic mice expressing Cre-recombinase under the control of the Pf4-Cre promoter to obtain Pdk1fl/flpf4-Cre+ (Pdk1−/−) and Pdk1fl/flpf4-Cre− (Pdk1fl/fl) littermates. Real-time polymerase chain reaction analysis revealed that Pdk1 was completely abrogated in Pdk1−/− MKs on transcript level, whereas Pdk1 transcription in other tissues was unaffected (Figure 1A). The complete loss of PDK1 on the protein level (molecular weight 63 kDa) in BM MKs derived from Pdk1−/− mice was confirmed by immunoblotting (Figure 1A; supplemental Figure 1).
![MK-specific PDK1 deficiency results in macrothrombocytopenia and a reduced platelet lifespan. (A) Arithmetic means ± standard error of the mean (SEM; n = 6, unpaired 2-tailed Student t test) of Pdk1 transcript levels in platelets and kidneys from Pdk1fl/fl and Pdk1−/− mice. Representative immunoblot of PDK1 protein levels in MKs from Pdk1fl/fl and Pdk1−/− mice (n = 5). mRNA, messenger RNA. (B) Arithmetic means ± SEM of platelet count and mean platelet volume of Pdk1fl/fl and Pdk1−/− mice aged 8 weeks (n = 13), 12 weeks (n = 22), and 18 weeks (n = 10). (C) Arithmetic means ± SEM (n = 12) of mean forward scatter (FSC) signal of Pdk1fl/fl and Pdk1−/− platelets. (D) Arithmetic means ± SEM (n = 8, unpaired 2-tailed Student t test) of relative spleen weight in Pdk1fl/fl and Pdk1−/− mice. (E) TPO levels of Pdk1fl/fl and Pdk1−/− mice. Arithmetic means ± SEM (n = 6, unpaired 2-tailed Student t test) are shown. TPO receptor/c-Mpl protein level in Pdk1fl/fl and Pdk1−/− MKs. Actin was used as loading control. Arithmetic means ± SEM (n = 5, unpaired 2-tailed Student t test) are shown. (F) Platelet counts of Pdk1fl/fl and Pdk1−/− mice before and 4 days after treatment with TPO (2 µg per animal per day). Arithmetic means ± SEM (n = 8, 2-way analysis of variance [ANOVA] with Bonferroni correction for multiple comparisons) are shown. (G) Endogenous survival of Pdk1fl/fl and Pdk1−/− platelets measured by determination of percent fluorescently labeled platelets in vivo at indicated time points after injection of DyLight 488 α-glycoprotein IX (GPIX) (n = 6, unpaired 2-tailed Student t test). (H) Platelet depletion in Pdk1fl/fl and Pdk1−/− mice was induced by IV injection of rat anti-GPIbα antibody (Emfret Analytics). Peripheral platelet counts were determined over a time course of 7 days by flow cytometry. The rate of platelet count recovery as compared with the respective initial platelet count is depicted (n = 4, unpaired 2-tailed Student t test). *P < .05 and **P < .01 as well as ##P < .01 indicate statistically significant differences.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/134/21/10.1182_blood.2019000185/7/m_bloodbld2019000185f1.png?Expires=1766276477&Signature=mwLHZ3mlgKS0YaHNZbUogFcunCNA4CkY9mv7zawhdsgOxTD-aPDgxEgR3WFdjyjAbGjEQDXSqdjxQ~gp1xcJ1oMw6sjPrh0b~mXW2Me856usgCsJyd40U-QyJZkpWB9IdsAfOrCw4gLl4~IT987lyrX7HXwni7X8tYim960yDhHh9BXqPo~FQ0BHb-dVyegT0ZQ~o9sGnmxToROb7CfExdUxzgBAz~jVAPw0lfi-DyYRuAMk4GABqD3KYMLiJMiTDveIib2PuZAMlgdWFL9XgTARCG8pliv6EHfMAeH5WykMMacqyj9~ND9rKZ~UUCMp~kc4nUcSB656uSqQ4hcFWQ__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
MK-specific PDK1 deficiency results in macrothrombocytopenia and a reduced platelet lifespan. (A) Arithmetic means ± standard error of the mean (SEM; n = 6, unpaired 2-tailed Student t test) of Pdk1 transcript levels in platelets and kidneys from Pdk1fl/fl and Pdk1−/− mice. Representative immunoblot of PDK1 protein levels in MKs from Pdk1fl/fl and Pdk1−/− mice (n = 5). mRNA, messenger RNA. (B) Arithmetic means ± SEM of platelet count and mean platelet volume of Pdk1fl/fl and Pdk1−/− mice aged 8 weeks (n = 13), 12 weeks (n = 22), and 18 weeks (n = 10). (C) Arithmetic means ± SEM (n = 12) of mean forward scatter (FSC) signal of Pdk1fl/fl and Pdk1−/− platelets. (D) Arithmetic means ± SEM (n = 8, unpaired 2-tailed Student t test) of relative spleen weight in Pdk1fl/fl and Pdk1−/− mice. (E) TPO levels of Pdk1fl/fl and Pdk1−/− mice. Arithmetic means ± SEM (n = 6, unpaired 2-tailed Student t test) are shown. TPO receptor/c-Mpl protein level in Pdk1fl/fl and Pdk1−/− MKs. Actin was used as loading control. Arithmetic means ± SEM (n = 5, unpaired 2-tailed Student t test) are shown. (F) Platelet counts of Pdk1fl/fl and Pdk1−/− mice before and 4 days after treatment with TPO (2 µg per animal per day). Arithmetic means ± SEM (n = 8, 2-way analysis of variance [ANOVA] with Bonferroni correction for multiple comparisons) are shown. (G) Endogenous survival of Pdk1fl/fl and Pdk1−/− platelets measured by determination of percent fluorescently labeled platelets in vivo at indicated time points after injection of DyLight 488 α-glycoprotein IX (GPIX) (n = 6, unpaired 2-tailed Student t test). (H) Platelet depletion in Pdk1fl/fl and Pdk1−/− mice was induced by IV injection of rat anti-GPIbα antibody (Emfret Analytics). Peripheral platelet counts were determined over a time course of 7 days by flow cytometry. The rate of platelet count recovery as compared with the respective initial platelet count is depicted (n = 4, unpaired 2-tailed Student t test). *P < .05 and **P < .01 as well as ##P < .01 indicate statistically significant differences.
MK-specific PDK1 deficiency results in macrothrombocytopenia and a reduced platelet lifespan. (A) Arithmetic means ± standard error of the mean (SEM; n = 6, unpaired 2-tailed Student t test) of Pdk1 transcript levels in platelets and kidneys from Pdk1fl/fl and Pdk1−/− mice. Representative immunoblot of PDK1 protein levels in MKs from Pdk1fl/fl and Pdk1−/− mice (n = 5). mRNA, messenger RNA. (B) Arithmetic means ± SEM of platelet count and mean platelet volume of Pdk1fl/fl and Pdk1−/− mice aged 8 weeks (n = 13), 12 weeks (n = 22), and 18 weeks (n = 10). (C) Arithmetic means ± SEM (n = 12) of mean forward scatter (FSC) signal of Pdk1fl/fl and Pdk1−/− platelets. (D) Arithmetic means ± SEM (n = 8, unpaired 2-tailed Student t test) of relative spleen weight in Pdk1fl/fl and Pdk1−/− mice. (E) TPO levels of Pdk1fl/fl and Pdk1−/− mice. Arithmetic means ± SEM (n = 6, unpaired 2-tailed Student t test) are shown. TPO receptor/c-Mpl protein level in Pdk1fl/fl and Pdk1−/− MKs. Actin was used as loading control. Arithmetic means ± SEM (n = 5, unpaired 2-tailed Student t test) are shown. (F) Platelet counts of Pdk1fl/fl and Pdk1−/− mice before and 4 days after treatment with TPO (2 µg per animal per day). Arithmetic means ± SEM (n = 8, 2-way analysis of variance [ANOVA] with Bonferroni correction for multiple comparisons) are shown. (G) Endogenous survival of Pdk1fl/fl and Pdk1−/− platelets measured by determination of percent fluorescently labeled platelets in vivo at indicated time points after injection of DyLight 488 α-glycoprotein IX (GPIX) (n = 6, unpaired 2-tailed Student t test). (H) Platelet depletion in Pdk1fl/fl and Pdk1−/− mice was induced by IV injection of rat anti-GPIbα antibody (Emfret Analytics). Peripheral platelet counts were determined over a time course of 7 days by flow cytometry. The rate of platelet count recovery as compared with the respective initial platelet count is depicted (n = 4, unpaired 2-tailed Student t test). *P < .05 and **P < .01 as well as ##P < .01 indicate statistically significant differences.
Mice with Pdk1-deficient MK/platelet lineage were viable and fertile, but displayed a significant and tendentially progressive macrothrombocytopenia (Figure 1B) at the ages of 8, 12, and 18 weeks without obvious signs of spontaneous hemorrhage. Analysis of the platelet forward scatter by flow cytometry confirmed the significantly increased size of Pdk1−/− platelets (Figure 1C). Because MK proliferation and consecutive thrombopoiesis are primarily triggered by the cytokine TPO, which binds to its MK surface receptor c-Mpl, we examined the TPO signaling in mice with MK-specific Pdk1 deficiency. As illustrated in Figure 1E, neither the TPO plasma levels nor the expression of the TPO receptor c-Mpl in MKs was significantly different in Pdk1−/− mice as compared with wild-type mice. Notably, exogenous TPO failed to sufficiently elevate platelet counts of Pdk1−/− mice to prevent the thrombocytopenia (Figure 1F). Although TPO injection was followed by significant elevations in platelet counts of wild-type mice after 96 hours, the platelet count of Pdk1−/− mice was not significantly increased, indicating that the TPO signaling in MKs includes a PDK1-dependent pathway. However, neither Akt phosphorylation nor apoptosis rate (as reflected by caspase activity and Bad phosphorylation) was significantly affected in MKs from Pdk1−/− mice as compared with wild-type MKs (supplemental Figure 2). To elucidate whether the macrothrombocytopenia in Pdk1−/− mice was caused by enhanced platelet turnover, the lifespan of circulating platelets was determined in Pdk1−/− mice and wild-type littermates. Clearance of Pdk1-deficient platelets was only slightly accelerated (Figure 1G), which may contribute to, but does not explain, the pronouncedly reduced platelet counts of Pdk1−/− mice. Moreover, the recovery of platelet counts after platelet depletion in vivo followed comparable kinetics in Pdk1fl/fl and Pdk1−/− mice, resulting in a sufficient restoration of the respective initial platelet levels within 7 days after platelet depletion (Figure 1H).
For a further examination of the origin of the observed impaired thrombopoiesis, BM megakaryopoiesis was investigated by anti-GPIbα immunostaining of BM sections from Pdk1−/− mice and wild-type littermates. As shown in Figure 2A, the BM of Pdk1−/− mice exhibited a remarkable MK hyperplasia reflected by a significantly increased number of GPIb-positive MKs within the BM without obvious signs of increased MK fragmentation. The spleen acts as a site of platelet production if BM-derived thrombopoiesis is not sufficient. Therefore, we analyzed spleen sections of Pdk1−/− mice and wild-type littermates for extramedullary megakaryopoiesis using GPIb immunohistological analysis as well as hematoxylin and eosin staining. Although only a mild splenomegaly was observed in Pdk1−/− mice (Figure 1D), we found a drastically increased extramedullary thrombopoiesis in Pdk1−/− mice as compared with wild-type mice as evident by the strong enrichment with GPIb-positive MKs (Figure 2B). However, as illustrated in supplemental Figure 3, Pdk1−/− mice displayed no significant modification of the hematopoietic stem and progenitor cell (HSPC) compartment in BM or spleen.
![Pdk1−/−mice display increased localization of MKs in the BM hematopoietic compartment with less sinusoidal contact, a substantial BM MK hyperplasia, and an upregulation of extramedullary thrombopoiesis. (A) Representative images of GPIb immunostaining of BM sections from 2-month-old Pdk1fl/fl and Pdk1−/− mice (left). Quantification of Pdk1fl/fl and Pdk1−/− BM MKs per visual field. Arithmetic means ± SEM (n = 6, unpaired 2-tailed Student t test; right) are shown. Scale bar equals 50 μm. (B) Representative images of hematoxylin and eosin stainings as well as GPIb immunostaining of spleen sections from 6-month-old Pdk1fl/fl and Pdk1−/− mice (left). Quantification of Pdk1fl/fl and Pdk1−/− spleen MKs per visual field. Arithmetic means ± SEM (n = 6, unpaired 2-tailed Student t test; right) are shown. Scale bar equals 50 μm. (C) Representative confocal microscopy images of immunostained BM sections (n = 5). Green, MKs (GPIb); red, sinusoids (endoglin, CD105); gray, nuclei (4′,6-diamidino-2-phenylindole [DAPI]). Scale bar equals 20 μm. Boxes in the left panels mark the section in the illustrations which are highlighted in a higher magnification in the right panels. (D) Arithmetic means ± SEM (n = 6, unpaired 2-tailed Student t test) of MKs per visual field in immunostained BM sections of Pdk1fl/fl and Pdk1−/− mice. (E) Quantification of MK distance from sinusoids in BM sections of Pdk1fl/fl and Pdk1−/− mice (means ± SEM, n = 6, 750 MKs, Wilcoxon-Mann-Whitney test). (F) Quantification of MK localization in the BM of Pdk1fl/fl and Pdk1−/− mice (arithmetic means ± SEM, n = 6, 2-way ANOVA with Bonferroni correction for multiple comparisons). BMHC, bone marrow hematopoietic compartment; Intrasin., intrasinusoidal; SC, sinusoidal contact. **P < .01 indicates statistically significant difference.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/134/21/10.1182_blood.2019000185/7/m_bloodbld2019000185f2.png?Expires=1766276477&Signature=LlIh5wqgdKUDtXbx25QQCk34qpaGiHpfAPyhTGhJJOZMteLiYrghCxqX-epxyGnZF3UkjdDQU4flxKGprzaN01EMFEcvBFv0B4hIz4CNTW5L~~3rAfAc9eLHNNd3cMWosOxpbPfcwNu7uq9TCRQa7jPge2~7LGGBLUdjscC8JdKbiiN8XT4iJZEwGxs1b3Ob~TuyP6ar3T4HbyyJb~xm32QsEYA0sFVDEA01IESjP7mGIPN21XEHcoDXMHjpHGNX2fx4lAdFXSX9siDUwwMwJwM2dGb9jllUU-hfDCwnd09ACCXq78c65kvXTqno4BGsAZprFFsJv4sLNDe0O9pYPQ__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Pdk1−/−mice display increased localization of MKs in the BM hematopoietic compartment with less sinusoidal contact, a substantial BM MK hyperplasia, and an upregulation of extramedullary thrombopoiesis. (A) Representative images of GPIb immunostaining of BM sections from 2-month-old Pdk1fl/fl and Pdk1−/− mice (left). Quantification of Pdk1fl/fl and Pdk1−/− BM MKs per visual field. Arithmetic means ± SEM (n = 6, unpaired 2-tailed Student t test; right) are shown. Scale bar equals 50 μm. (B) Representative images of hematoxylin and eosin stainings as well as GPIb immunostaining of spleen sections from 6-month-old Pdk1fl/fl and Pdk1−/− mice (left). Quantification of Pdk1fl/fl and Pdk1−/− spleen MKs per visual field. Arithmetic means ± SEM (n = 6, unpaired 2-tailed Student t test; right) are shown. Scale bar equals 50 μm. (C) Representative confocal microscopy images of immunostained BM sections (n = 5). Green, MKs (GPIb); red, sinusoids (endoglin, CD105); gray, nuclei (4′,6-diamidino-2-phenylindole [DAPI]). Scale bar equals 20 μm. Boxes in the left panels mark the section in the illustrations which are highlighted in a higher magnification in the right panels. (D) Arithmetic means ± SEM (n = 6, unpaired 2-tailed Student t test) of MKs per visual field in immunostained BM sections of Pdk1fl/fl and Pdk1−/− mice. (E) Quantification of MK distance from sinusoids in BM sections of Pdk1fl/fl and Pdk1−/− mice (means ± SEM, n = 6, 750 MKs, Wilcoxon-Mann-Whitney test). (F) Quantification of MK localization in the BM of Pdk1fl/fl and Pdk1−/− mice (arithmetic means ± SEM, n = 6, 2-way ANOVA with Bonferroni correction for multiple comparisons). BMHC, bone marrow hematopoietic compartment; Intrasin., intrasinusoidal; SC, sinusoidal contact. **P < .01 indicates statistically significant difference.
Pdk1−/−mice display increased localization of MKs in the BM hematopoietic compartment with less sinusoidal contact, a substantial BM MK hyperplasia, and an upregulation of extramedullary thrombopoiesis. (A) Representative images of GPIb immunostaining of BM sections from 2-month-old Pdk1fl/fl and Pdk1−/− mice (left). Quantification of Pdk1fl/fl and Pdk1−/− BM MKs per visual field. Arithmetic means ± SEM (n = 6, unpaired 2-tailed Student t test; right) are shown. Scale bar equals 50 μm. (B) Representative images of hematoxylin and eosin stainings as well as GPIb immunostaining of spleen sections from 6-month-old Pdk1fl/fl and Pdk1−/− mice (left). Quantification of Pdk1fl/fl and Pdk1−/− spleen MKs per visual field. Arithmetic means ± SEM (n = 6, unpaired 2-tailed Student t test; right) are shown. Scale bar equals 50 μm. (C) Representative confocal microscopy images of immunostained BM sections (n = 5). Green, MKs (GPIb); red, sinusoids (endoglin, CD105); gray, nuclei (4′,6-diamidino-2-phenylindole [DAPI]). Scale bar equals 20 μm. Boxes in the left panels mark the section in the illustrations which are highlighted in a higher magnification in the right panels. (D) Arithmetic means ± SEM (n = 6, unpaired 2-tailed Student t test) of MKs per visual field in immunostained BM sections of Pdk1fl/fl and Pdk1−/− mice. (E) Quantification of MK distance from sinusoids in BM sections of Pdk1fl/fl and Pdk1−/− mice (means ± SEM, n = 6, 750 MKs, Wilcoxon-Mann-Whitney test). (F) Quantification of MK localization in the BM of Pdk1fl/fl and Pdk1−/− mice (arithmetic means ± SEM, n = 6, 2-way ANOVA with Bonferroni correction for multiple comparisons). BMHC, bone marrow hematopoietic compartment; Intrasin., intrasinusoidal; SC, sinusoidal contact. **P < .01 indicates statistically significant difference.
The combination of increased MK numbers in the BM, along with decreased platelet counts and a compensatory upregulation of extramedullary megakaryopoiesis, suggested that lack of Pdk1 affected platelet production in the BM. To further elucidate the functional consequence of Pdk1 deficiency for platelet biogenesis, we visualized MK localization and morphology in immunostained BM cryosections of intact murine femora. The visualization of the MK distribution within the entire femora confirmed an increased number of BM MKs in Pdk1−/− mice, but did not indicate obvious alterations in their overall distribution in the BM as compared with the control (supplemental Figure 4). Detailed analyses revealed that in wild-type mice, the majority of GPIX-positive MKs was in close contact with BM sinusoids stained with marker endoglin (Figure 2C). In contrast, in Pdk1−/− mice, the number of MKs with direct contact to sinusoids was significantly reduced (Figure 2C). Although Pdk1 deficiency was accompanied by a significant increase of total number of BM MKs (Figure 2D), Pdk1−/− MKs were located further away from the sinusoids (Figure 2E), resulting in a reduced percentage of MKs with direct sinusoidal contact and conversely an accumulation of MKs in the BM hematopoietic compartment (Figure 2E-F), indicating insufficient transendothelial platelet biogenesis.
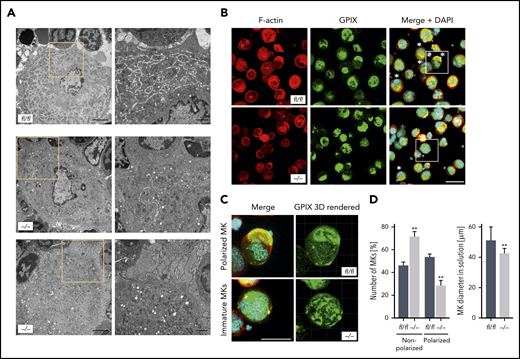
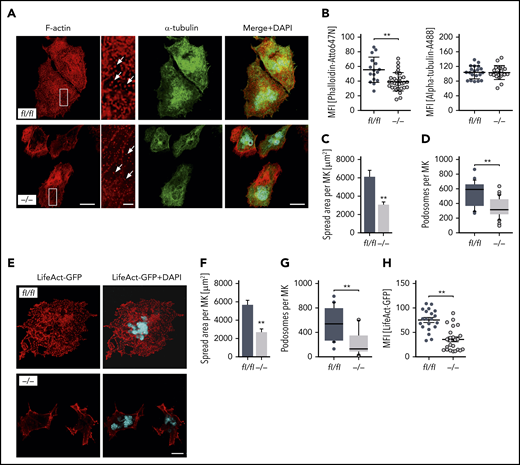
Given that MK localization and polarization at sinusoids are critical for directed platelet biogenesis,8,9 we next analyzed the morphology of the DMS as well as the polarization capacity of Pdk1-deficient BM MKs. The ultrastructure of MKs in BM sections of Pdk1−/− and Pdk1fl/fl mice was investigated by TEM. Although wild-type MKs displayed characteristic membrane invaginations, the DMS was underdeveloped in MKs of Pdk1−/− mice as reflected by fewer invaginations in the periphery and around the nucleus (Figure 3A). By using immunofluorescence confocal microscopy with consecutive 3-dimensional surface rendering, we determined DMS polarization in cultured MKs, a process critically dependent on actin reorganization. As pointed out in Figure 3B-D and supplemental videos 1 and 2, a significantly higher number of MKs with Pdk1 deletion showed a nonpolarized DMS as compared with wild-type MKs (46.35% ± 2.58% vs 71.5% ± 4.62%, mean ± standard deviation [SD]; Figure 3D left). Furthermore, the diameter of cultured Pdk1−/− MKs was significantly reduced (Figure 3D right). To address the question of whether a defect in actin cytoskeleton rearrangements may underlie the impaired DMS development and polarization of Pdk1−/− MKs, we further examined the cytoskeletal structure and composition of cultured Pdk1−/− and wild-type BM MKs adherent to Horm collagen (which mainly contains collagen type I) by immunofluorescence confocal microscopy (Figure 4A), including LifeAct green fluorescent protein (GFP) imaging (Figure 4E). In Pdk1−/− MKs, assembly of F-actin was disturbed (Figure 4A-E) as reflected by regions showing accumulation of F-actin, whereas, overall, the mean fluorescence intensity (MFI) of phalloidin-Atto647N (Figure 4B, left) and LifeAct-GFP (Figure 4H) was significantly decreased. Notably, the organization of the tubulin cytoskeleton was not obviously altered in Pdk1−/− compared with wild-type MKs (Figure 4A-B, right), supporting the observation that Pdk1 is a regulator of actin rather than tubulin dynamics in MKs. As a further consequence of defective actin cytoskeletal dynamics, the spreading area was significantly diminished in Pdk1−/− MKs as compared with Pdk1fl/fl MKs (Figure 4C). During thrombopoiesis, MK podosomes, actin-based structures, have been suggested to be important for MK interactions with matrix proteins and proplatelet penetration through the basement membrane.28 Strikingly, podosomes, as identified by their characteristic F-actin staining, were smaller, and their number was significantly reduced in Pdk1−/− MKs as compared with Pdk1fl/fl MKs adherent to collagen I (Figure 4A,D,E,G). Because formation of podosomes with F-actin cores in MKs is mainly driven by actin polymerization via WASp and the Arp2/3 complex,28 we further performed immunofluorescence stainings for WASp and Arp2 to highlight the impaired formation of podosomes in Pdk1−/− MKs spread on collagen (supplemental Figure 5).
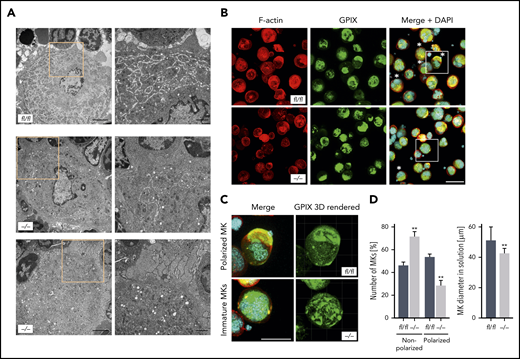
Pdk1-deficient BM MKs show an abnormal ultrastructure with disrupted DMS and abrogated MK polarization. (A) Representative TEM images of Pdk1fl/fl (upper) and Pdk1−/− (lower) MKs in the BM showing decreased presence of demarcation membranes and large granula-free zones upon Pdk1 deficiency. Overview (left; scale bar equals 3 μm). Detail (right; scale bar equals 1 μm). Boxes mark the section in the illustrations which are highlighted in a higher magnification in the right panels. (B) Representative confocal images (maximum projection) of BM-derived Pdk1fl/fl and Pdk1−/− MKs cultured in vitro for 5 days and stained for F-actin (Phalloidin-Atto647N, red), nuclei (DAPI, blue), and GPIX (GPIX-Alexa488, green). Three-dimensional surface rendering of respective z-stacks (right; Imaris 9.2). White asterisks indicate polarized MKs. Scale bar equals 40 μm. Boxes mark the section in the illustrations which are highlighted in a higher magnification in panel C. (C) Detailed visualization of DMS polarization of individual cultured Pdk1fl/fl and Pdk1−/− BM MKs. (D) Quantification of the percent of polarized and nonpolarized Pdk1fl/fl and Pdk1−/− BM MKs (left) and the MK diameter (right). Arithmetic means ± SD (n = 6 mice, at least 100 MKs per genotype were analyzed, unpaired 2-tailed Student t test). **P < .01 indicates statistically significant difference.
Pdk1-deficient BM MKs show an abnormal ultrastructure with disrupted DMS and abrogated MK polarization. (A) Representative TEM images of Pdk1fl/fl (upper) and Pdk1−/− (lower) MKs in the BM showing decreased presence of demarcation membranes and large granula-free zones upon Pdk1 deficiency. Overview (left; scale bar equals 3 μm). Detail (right; scale bar equals 1 μm). Boxes mark the section in the illustrations which are highlighted in a higher magnification in the right panels. (B) Representative confocal images (maximum projection) of BM-derived Pdk1fl/fl and Pdk1−/− MKs cultured in vitro for 5 days and stained for F-actin (Phalloidin-Atto647N, red), nuclei (DAPI, blue), and GPIX (GPIX-Alexa488, green). Three-dimensional surface rendering of respective z-stacks (right; Imaris 9.2). White asterisks indicate polarized MKs. Scale bar equals 40 μm. Boxes mark the section in the illustrations which are highlighted in a higher magnification in panel C. (C) Detailed visualization of DMS polarization of individual cultured Pdk1fl/fl and Pdk1−/− BM MKs. (D) Quantification of the percent of polarized and nonpolarized Pdk1fl/fl and Pdk1−/− BM MKs (left) and the MK diameter (right). Arithmetic means ± SD (n = 6 mice, at least 100 MKs per genotype were analyzed, unpaired 2-tailed Student t test). **P < .01 indicates statistically significant difference.
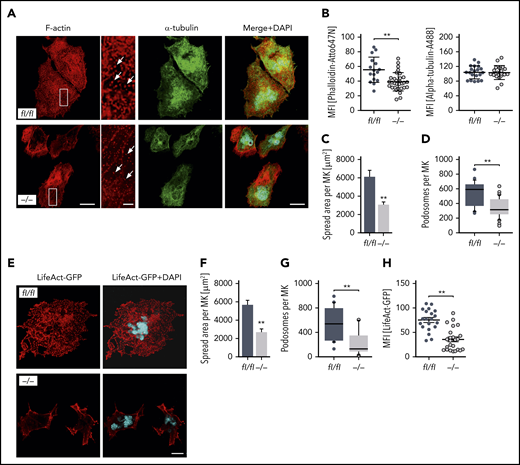
MK-specific deletion of Pdk1 results in defective actin polymerization with disrupted F-actin assembly, impaired spreading, as well as reduced podosome number and morphology upon adhesion on collagen. (A) Representative confocal images (maximum projection) of BM-derived Pdk1fl/fl and Pdk1−/− MKs adherent to collagen (50 μg/mL) and stained for F-actin and α-tubulin displaying a disrupted F-actin organization in the cytoskeleton of Pdk1−/− MKs with impaired spreading and a significant reduction of podosome density. Phalloidin-Atto647N staining was used for F-actin (red); α-tubulin-A488 staining was used for α-tubulin (green), and DAPI was used for nuclei staining (blue). White arrows indicate podosomes. Scale bar equals 40 μm (overview) or 10 μm (detail). (B) Arithmetic means ± SD (left; n = 36 to 59, Wilcoxon-Mann-Whitney test) of F-actin expression (reflected by MFI of Phalloidin-Atto647N) in Pdk1fl/fl and Pdk1−/− MKs upon adhesion to collagen for 3 hours.Arithmetic means ± SD (right; n = 40 to 48, Wilcoxon-Mann-Whitney test) of α-tubulin expression (reflected by MFI of anti–α-tubulin-Alexa488 antibody) in Pdk1fl/fl and Pdk1−/− MKs. (C) Arithmetic means ± SD (n = 74 to 86, unpaired 2-tailed Student t test) of Pdk1fl/fl and Pdk1−/− MK spreading area on collagen. (D) Means ± SD (n = 38 to 57, Wilcoxon-Mann-Whitney test) of podosome number in BM-derived MKs of Pdk1fl/fl and Pdk1−/− mice. (E) Representative confocal images of BM-derived Pdk1fl/fl and Pdk1−/− MKs adherent to collagen (50 μg/mL) and stained for polymerized F-actin using LifeAct-TagGFP2 (red). DAPI (blue) was used for nuclei staining. Scale bar equals 20 μm. (F) Arithmetic means ± SEM (n = 42 to 51, unpaired 2-tailed Student t test) of Pdk1fl/fl and Pdk1−/− MK spreading area on collagen. (G) Means ± SEM (n = 42 to 51, Wilcoxon-Mann-Whitney test) of podosome number in MKs derived from BM of Pdk1fl/fl and Pdk1−/− mice adherent on collagen (related to panel E). (H) Arithmetic means ± SEM (n = 20 to 23, Wilcoxon-Mann-Whitney test) of F-actin expression (reflected by MFI of LifeAct-TagGFP2) in Pdk1fl/fl and Pdk1−/− MKs upon adhesion to collagen for 3 hours. **P < .01 indicates statistically significant difference.
MK-specific deletion of Pdk1 results in defective actin polymerization with disrupted F-actin assembly, impaired spreading, as well as reduced podosome number and morphology upon adhesion on collagen. (A) Representative confocal images (maximum projection) of BM-derived Pdk1fl/fl and Pdk1−/− MKs adherent to collagen (50 μg/mL) and stained for F-actin and α-tubulin displaying a disrupted F-actin organization in the cytoskeleton of Pdk1−/− MKs with impaired spreading and a significant reduction of podosome density. Phalloidin-Atto647N staining was used for F-actin (red); α-tubulin-A488 staining was used for α-tubulin (green), and DAPI was used for nuclei staining (blue). White arrows indicate podosomes. Scale bar equals 40 μm (overview) or 10 μm (detail). (B) Arithmetic means ± SD (left; n = 36 to 59, Wilcoxon-Mann-Whitney test) of F-actin expression (reflected by MFI of Phalloidin-Atto647N) in Pdk1fl/fl and Pdk1−/− MKs upon adhesion to collagen for 3 hours.Arithmetic means ± SD (right; n = 40 to 48, Wilcoxon-Mann-Whitney test) of α-tubulin expression (reflected by MFI of anti–α-tubulin-Alexa488 antibody) in Pdk1fl/fl and Pdk1−/− MKs. (C) Arithmetic means ± SD (n = 74 to 86, unpaired 2-tailed Student t test) of Pdk1fl/fl and Pdk1−/− MK spreading area on collagen. (D) Means ± SD (n = 38 to 57, Wilcoxon-Mann-Whitney test) of podosome number in BM-derived MKs of Pdk1fl/fl and Pdk1−/− mice. (E) Representative confocal images of BM-derived Pdk1fl/fl and Pdk1−/− MKs adherent to collagen (50 μg/mL) and stained for polymerized F-actin using LifeAct-TagGFP2 (red). DAPI (blue) was used for nuclei staining. Scale bar equals 20 μm. (F) Arithmetic means ± SEM (n = 42 to 51, unpaired 2-tailed Student t test) of Pdk1fl/fl and Pdk1−/− MK spreading area on collagen. (G) Means ± SEM (n = 42 to 51, Wilcoxon-Mann-Whitney test) of podosome number in MKs derived from BM of Pdk1fl/fl and Pdk1−/− mice adherent on collagen (related to panel E). (H) Arithmetic means ± SEM (n = 20 to 23, Wilcoxon-Mann-Whitney test) of F-actin expression (reflected by MFI of LifeAct-TagGFP2) in Pdk1fl/fl and Pdk1−/− MKs upon adhesion to collagen for 3 hours. **P < .01 indicates statistically significant difference.
Based on these results, we hypothesize that defective actin dynamics and the subsequently disturbed DMS formation, polarization, and podosome formation of MKs with a genetic knockout of Pdk1 result in an insufficient transendothelial proplatelet formation and consequently the significant thrombocytopenia observed in Pdk1−/− mice. Notably, on fibrinogen-coated surfaces, cytoskeletal structure and spreading area of Pdk1-deficient MKs were comparable to wild-type MKs (supplemental Figure 6).
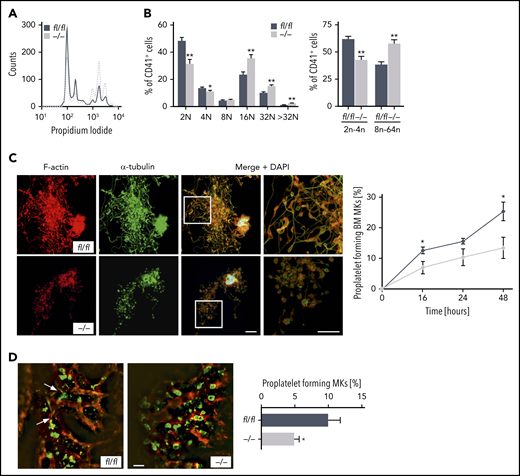
Furthermore, alterations of the actin cytoskeletal network in maturating MKs are critical to polyploidization.29 Thus, we next examined whether Pdk1 deficiency may affect MK ploidy and proplatelet formation. As revealed by analyses of DNA content analyses in CD41+ MKs flushed out of BM, we found that deletion of PDK1 markedly increased MK polyploidization because Pdk1−/− MKs showed significantly increased 16N, 32N, and >32N populations as well as a significantly higher percentage of MKs with a DNA content >8N (Figure 5A-B).
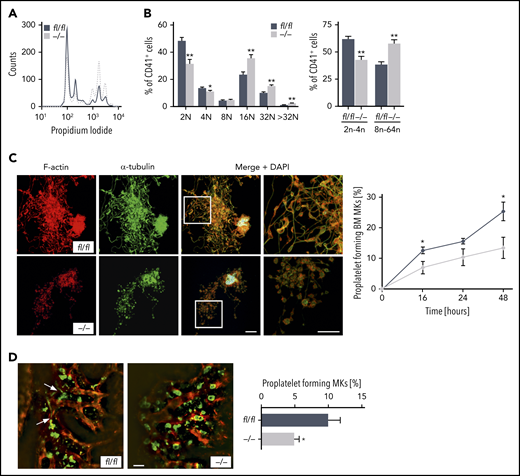
PDK1 is a critical regulator of MK ploidy and proplatelet formation in vitro and in vivo. (A) Representative ploidy histogram (n = 6) of Pdk1fl/fl MKs (black trace) and Pdk1−/− MKs is shown. (B) DNA content in Pdk1fl/fl and Pdk1−/− CD41+ BM-derived MKs cultured for 5 days (left). Arithmetic means ± SEM (n = 6, 2-way ANOVA with Bonferroni correction for multiple comparisons) are shown. Percentage of Pdk1fl/fl and Pdk1−/− CD41+ BM-derived MKs with a DNA content greater (right bars) and smaller (left bars) than 8N (right). Arithmetic means ± SEM (n = 6, 2-way ANOVA with Bonferroni correction for multiple comparisons) are shown. (C) Quantification of the percentage of proplatelet-forming BM-derived MKs from Pdk1fl/fl and Pdk1−/− mice in vitro. Representative immunostainings of proplatelet-forming MKs from Pdk1fl/fl (upper) and Pdk1−/− mice (lower) are shown (left). Red, F-actin (Phalloidin-Atto647N); green, α-tubulin (anti–α-tubulin-Alexa488), cyan, (DAPI) nuclei. Scale bar equals 40 μm. Pdk1fl/fl and Pdk1−/− MKs extending proplatelets were counted 16, 24, and 48 hours after bovine serum albumin gradient and expressed as percentage of total MKs per field of view (right). Arithmetic means ± SD (n = 3, unpaired 2-tailed Student t test). (D) 2P-IVM revealing reduced proplatelet formation in Pdk1−/− MKs in vivo. Platelets and MKs were stained with anti-GPIX antibody derivatives (green), and the vessel lumen was labeled using fluorescein isothiocyanate (FITC)–bovine serum albumin and anti-CD105 antibodies (red) (left). Representative median projections of 30-µm z-stacks from the BM of Pdk1fl/fl and Pdk1−/− MKs are depicted. Proplatelet-forming MKs (white arrows indicating proplatelets) were counted, and the ratio per mouse was assessed (arithmetic means ± SEM, n = 5 to 6 ice, 4 to 10 z-stacks per mouse, unpaired 2-sided Welch test; right). Scale bar equals 50 µm. *P < .05 and **P < .01 indicate statistically significant differences.
PDK1 is a critical regulator of MK ploidy and proplatelet formation in vitro and in vivo. (A) Representative ploidy histogram (n = 6) of Pdk1fl/fl MKs (black trace) and Pdk1−/− MKs is shown. (B) DNA content in Pdk1fl/fl and Pdk1−/− CD41+ BM-derived MKs cultured for 5 days (left). Arithmetic means ± SEM (n = 6, 2-way ANOVA with Bonferroni correction for multiple comparisons) are shown. Percentage of Pdk1fl/fl and Pdk1−/− CD41+ BM-derived MKs with a DNA content greater (right bars) and smaller (left bars) than 8N (right). Arithmetic means ± SEM (n = 6, 2-way ANOVA with Bonferroni correction for multiple comparisons) are shown. (C) Quantification of the percentage of proplatelet-forming BM-derived MKs from Pdk1fl/fl and Pdk1−/− mice in vitro. Representative immunostainings of proplatelet-forming MKs from Pdk1fl/fl (upper) and Pdk1−/− mice (lower) are shown (left). Red, F-actin (Phalloidin-Atto647N); green, α-tubulin (anti–α-tubulin-Alexa488), cyan, (DAPI) nuclei. Scale bar equals 40 μm. Pdk1fl/fl and Pdk1−/− MKs extending proplatelets were counted 16, 24, and 48 hours after bovine serum albumin gradient and expressed as percentage of total MKs per field of view (right). Arithmetic means ± SD (n = 3, unpaired 2-tailed Student t test). (D) 2P-IVM revealing reduced proplatelet formation in Pdk1−/− MKs in vivo. Platelets and MKs were stained with anti-GPIX antibody derivatives (green), and the vessel lumen was labeled using fluorescein isothiocyanate (FITC)–bovine serum albumin and anti-CD105 antibodies (red) (left). Representative median projections of 30-µm z-stacks from the BM of Pdk1fl/fl and Pdk1−/− MKs are depicted. Proplatelet-forming MKs (white arrows indicating proplatelets) were counted, and the ratio per mouse was assessed (arithmetic means ± SEM, n = 5 to 6 ice, 4 to 10 z-stacks per mouse, unpaired 2-sided Welch test; right). Scale bar equals 50 µm. *P < .05 and **P < .01 indicate statistically significant differences.
To therefore further study the consequence of Pdk1 deficiency in MKs for platelet biogenesis, we performed in vitro proplatelet formation assays using both BM-derived MKs (Figure 5C) and fetal liver cell–derived MKs (supplemental Figure 7). Consistent with the previous findings, proplatelet formation of Pdk1−/− MKs was significantly reduced, and visualization of individual proplatelets by F-actin and α-tubulin staining revealed reduced branching and increased size of swellings and tips as compared with wild-type MKs (Figure 5C). By using 2-photon intravital microscopy (2P-IVM), we visualized MKs and sinusoids in the BM in the mouse skull and analyzed proplatelet formation under steady-state conditions in vivo. According to 2P-IVM examinations, Pdk1−/− MKs were unable to efficiently release proplatelets into the vascular sinusoids in vivo. As illustrated in Figure 5D, only 4.99% ± 0.69% (mean ± SEM) of Pdk1-null MKs produced proplatelets in vivo compared with 10.08% ± 1.66% of wild-type MKs.
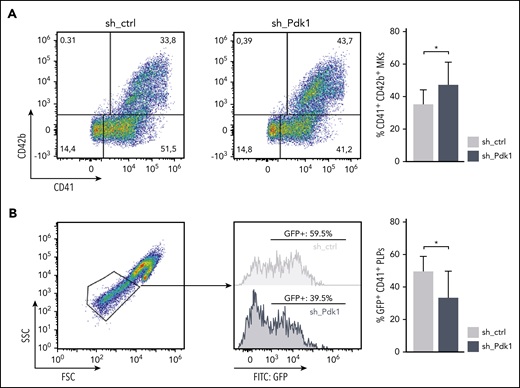
To assess reproducibility of our findings in the human system, we performed PDK1 short hairpin RNA (shRNA)-mediated knockdown on CD34+ cell-derived MKs. Knockdown efficiency of 3 different shRNAs was tested initially using the K562 cell line (supplemental Figure 8). The most efficient shRNA vector was chosen for transduction of human CD34+ HSPCs derived from 2 different donors in direct comparison with a nonsilencing shRNA. After 14 days of liquid culture, we observed markedly more mature MKs after PDK1 knockdown compared with controls, as indicated by elevated CD41/CD42b double-positive cell fractions (Figure 6A). Moreover, strikingly, depletion of PDK1 resulted in significantly decreased production of platelet-like particles (PLPs) (Figure 6B), thus independently corroborating our observations of Pdk1−/− mice in human cells.
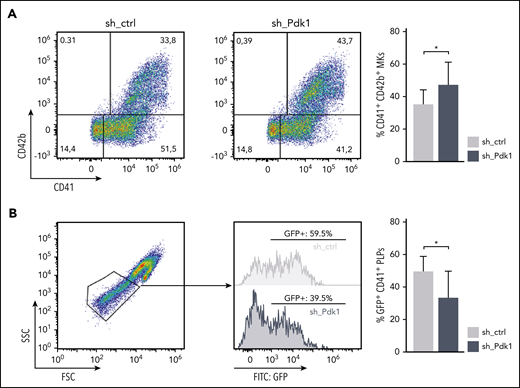
Increased maturation but decreased formation of PLPs in human CD34+-derived MKs upon knockdown of PDK1. (A) Human CD34+ HSPCs cultured for 14 days under MK differentiation conditions transduced with either an shRNA-bearing pGIPZ lentiviral vector (sh_PDK1) or a nonsilencing shRNA control (sh_ctrl). GFP served as a marker for positive transduction. Differentiation potency was quantified by flow cytometry using GPIIb (CD41) and GP1bα (CD42b) as markers for mature MKs. Representative pseudocolor plots of 1 donor with the respective indicated shRNA (left). Quantification of mature CD41/CD42b double-positive MKs assessed in 3 independent experiments with CD34+ cells from 2 different donors (right). Wilcoxon matched-pairs signed rank test was used to calculate statistical significance. (B) PLPs of day 14 MKs subgated from CD41-positive events. Representative scatter plot showing clearly defined PLPs (gate adjusted on peripheral blood PLPs) together with histograms indicating the amount of GFP-positive PLPs from 1 exemplarily chosen culture in each condition. Quantification of CD41-subgated GFP-positive PLPs in the PLP gate assessed in 3 independent experiments with CD34+ cells from 2 different donors (right). Wilcoxon matched-pairs signed rank test was used to calculate statistical significance. *P < .05 indicates statistically significant difference. SSC, side scatter.
Increased maturation but decreased formation of PLPs in human CD34+-derived MKs upon knockdown of PDK1. (A) Human CD34+ HSPCs cultured for 14 days under MK differentiation conditions transduced with either an shRNA-bearing pGIPZ lentiviral vector (sh_PDK1) or a nonsilencing shRNA control (sh_ctrl). GFP served as a marker for positive transduction. Differentiation potency was quantified by flow cytometry using GPIIb (CD41) and GP1bα (CD42b) as markers for mature MKs. Representative pseudocolor plots of 1 donor with the respective indicated shRNA (left). Quantification of mature CD41/CD42b double-positive MKs assessed in 3 independent experiments with CD34+ cells from 2 different donors (right). Wilcoxon matched-pairs signed rank test was used to calculate statistical significance. (B) PLPs of day 14 MKs subgated from CD41-positive events. Representative scatter plot showing clearly defined PLPs (gate adjusted on peripheral blood PLPs) together with histograms indicating the amount of GFP-positive PLPs from 1 exemplarily chosen culture in each condition. Quantification of CD41-subgated GFP-positive PLPs in the PLP gate assessed in 3 independent experiments with CD34+ cells from 2 different donors (right). Wilcoxon matched-pairs signed rank test was used to calculate statistical significance. *P < .05 indicates statistically significant difference. SSC, side scatter.
Together, these findings demonstrate that PDK1 is required for proplatelet formation and indicate that it functions as a critical regulator in the terminal steps of platelet biogenesis in both mice and humans.
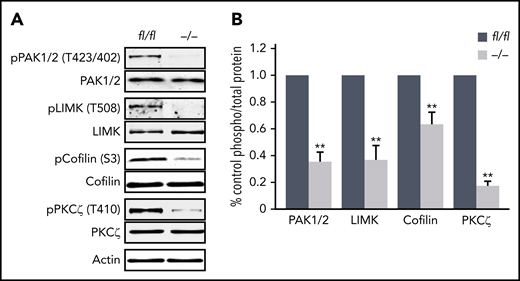
Finally, we aimed to identify potential mechanisms by which Pdk1 deletion may affect MK actin cytoskeleton organization and consecutively MK maturation and proplatelet formation. Previously, it has been shown that opposing signaling of the small Rho GTPases RhoA and Cdc42 is required for transendothelial proplatelet formation in the BM in vivo.9 However, the activity of these cytoskeleton-regulating GTPases was not affected in cultured Pdk1−/− MKs (supplemental Figure 9). Downstream targets of PDK1 include p21-activated kinase (PAK).30 Genetic knockout of PAK2 resulted in a restrained endomitosis during megakaryopoiesis and a substantial defect in the MK actin cytoskeleton dynamics with impaired platelet production due to an impaired activation of the PAK substrate LIM domain kinase (LIMK).29 As revealed by immunoblotting of enzymatically relevant phosphorylation sites of PAK1/2 (Thr423/402) and LIMK (Thr508) critical for activation of these kinases, we found significantly reduced levels of active PAK and LIMK in BM-derived cultured Pdk1−/− MKs (Figure 7A-B). Decreased expression of polymerized F-actin in the cytoskeleton of Pdk1-deficient MKs supports the hypothesis that Pdk1 deletion may increase cofilin activity as a result of impaired LIMK phosphorylation. Cofilin is active in a nonphosphorylated state, and its capacity to depolymerize F-actin is induced by phosphorylation on Ser3 by activated LIMK.19 Western blot analysis disclosed a significant reduction of cofilin phosphorylation (Ser3) in Pdk1−/− MKs with consecutively increased cofilin activity (Figure 7A-B), suggesting that Pdk1−/− MKs undergo a more frequent actin severing and depolymerization. Furthermore, interaction of PI3K signaling with atypical PKC was reported to determine cell polarity in neuronal cells31,32 and is potentially involved in polarization of MKs during thrombopoiesis.9 We thus investigated the phosphorylation state of atypical protein kinase C isoform ζ (PKCζ) in BM-derived Pdk1-null MKs and found that Pdk1 deficiency resulted in a significantly reduced phosphorylation of PKCζ at its activation site Thr410 (Figure 6A-B). These data establish PDK1 as an important regulator in the intracellular signaling in MKs underlying MK actin cytoskeleton dynamics and polarization during thrombopoiesis.
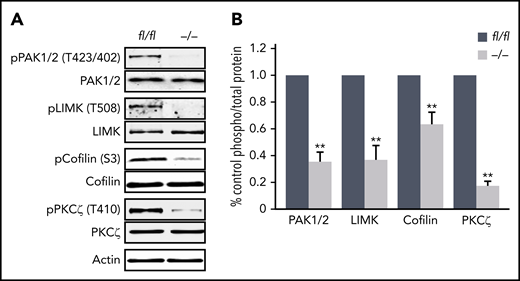
Abolished phosphorylation of proteins in Pdk1−/−MKs critical to MK actin cytoskeleton organization and polarization. (A) Representative immunoblot of phosphorylation levels of actin cytoskeleton regulating proteins PAK1/2 at Thr423/402, LIMK1 at Thr508, cofilin at Ser3, and atypical PKCζ at Thr401 in wild-type (Pdk1fl/fl) and Pdk1−/− MKs (n = 5). (B) Arithmetic means ± SEM (n = 5, unpaired 2-tailed Student t test) of blot densities actin cytoskeleton regulating proteins in wild-type (Pdk1fl/fl) and Pdk1−/− MKs quantified as ratio of phospho to total protein and calculated as percentage of control. **P < .01 indicates statistically significant differences.
Abolished phosphorylation of proteins in Pdk1−/−MKs critical to MK actin cytoskeleton organization and polarization. (A) Representative immunoblot of phosphorylation levels of actin cytoskeleton regulating proteins PAK1/2 at Thr423/402, LIMK1 at Thr508, cofilin at Ser3, and atypical PKCζ at Thr401 in wild-type (Pdk1fl/fl) and Pdk1−/− MKs (n = 5). (B) Arithmetic means ± SEM (n = 5, unpaired 2-tailed Student t test) of blot densities actin cytoskeleton regulating proteins in wild-type (Pdk1fl/fl) and Pdk1−/− MKs quantified as ratio of phospho to total protein and calculated as percentage of control. **P < .01 indicates statistically significant differences.
Discussion
Impaired platelet production appears in many hematological disorders and may result in thrombocytopenia with severe bleeding. Nevertheless, exact signaling pathways in MKs that are crucial for the process of thrombopoiesis and that are affected in disorders characterized by (macro-) thrombocytopenia still remain poorly defined. In the present study, we provide strong evidence that PDK1-dependent signaling plays an important role in platelet production. In detail, the major findings of this study are as follows: (i) MK lineage–specific PDK1 deletion results in significant macrothrombocytopenia with increased extramedullary megakaryopoiesis. (ii) Lack of Pdk1 leads to altered MK ultrastructure and increased polyploidization. (iii) PDK1 is a potent regulator of the MK actin cytoskeleton remodeling and podosome formation during MK maturation involving phosphorylation-dependent signaling via PAK/LIMK/cofilin. (iv) PDK1 determines phosphorylation of atypical PKCζ and is crucial for DMS polarization during thrombopoiesis. (v) Pdk1 deficiency in MKs resulted in diminished proplatelet formation without affecting MK apoptosis. (vi) Knockdown of PDK1 in human MKs resulted in increased maturity but reduced formation of PLPs.
PDK1 plays an essential role in neurodegenerative disorders33 as well as malignant tumors34 and acute myeloid leukemia,35 drives tumor invasion and metastasis,36 and is thus considered a potential target for antitumor and antileukemic therapy.37,38 Our current study has implications for the long-term therapeutic use of PDK1 inhibitors in patients with cancer and leukemia, which may result in a defective thrombopoiesis and may thus increase the risk of thrombocytopenia and bleeding disorders in those patients.
Genetic deletion of Pdk1 in the murine BM is associated with macrothrombocytopenia and altered MK ultrastructure as well as increased ploidy with defective proplatelet formation in vitro and in vivo. Importantly, our results indicate that PDK1 has a similar function in human megakaryopoiesis and thrombopoiesis, because knockdown of PDK1 in CD34+-differentiated MKs increased the number of mature MKs while at the same time reducing the capacity to produced PLPs. Because PDK1 plays essential roles in cell growth, proliferation, and survival, mainly by activating Akt-dependent signaling, it has already been speculated that PDK1 may regulate MK apoptosis during proplatelet formation.27 However, we found neither a significant effect on expression or activation of Akt nor an impact on pro- or antiapoptotic signaling cascades in MKs because caspase activity, BAD phosphorylation, or Annexin-V exposure was comparable in Pdk1−/− and wild-type MKs (supplemental Figure 2), altogether suggesting that PDK1 is dispensable for the apoptotic pathways in MKs. PDK2 has been described as an equivalent of the mTORC2 complex,39,40 and besides PDK1, the mTORC2 complex is needed to phosphorylate Akt at Ser473.41 Because neither mTOR expression nor Akt phosphorylation at Ser473 was different in Pdk1−/− MKs compared with wild-type MKs (supplemental Figure 10), intact Akt signaling via PDK2/mTORC2 could be an explanation for the unaffected BAD phosphorylation and caspase activity in Pdk1−/− MKs.
Platelet lifespan and the recovery of platelet counts after platelet depletion in vivo were only slightly affected, whereas stimulation of MK with TPO failed to increase the rate of platelet production and thereby the platelet count in Pdk1-deficient mice. These results are in line with findings by Chen et al that Pdk1 deficiency did not enhance platelet apoptosis,22 but may play a substantial role in the regulation of MK maturation and proplatelet formation. During MK maturation, MKs undergo cytoskeletal alteration critical for polyploidy and proplatelet formation.29 Microtubules assembled from α- and β-tubulin heterodimers are the driving force of proplatelet elongation.1,3 Actin polymerization and filament turnover are essential for maintaining reorganization of MK cytoskeleton, underlying DMS formation and MK polarization during proplatelet formation, proplatelet branching, and subsequent platelet production.7,8,42 Although structure and expression of tubulin cytoskeleton were unaffected, Pdk1 deficiency resulted in a significantly aberrant F-actin organization in BM-derived MKs. In line with previous findings that proper DMS organization relies on efficient F-actin dynamics to polarize MK membranes and nuclei during thrombopoiesis, we found a disrupted DMS structure with a consecutively impaired polarization of Pdk1-deficient MKs as a functional consequence of the defective F-actin network. The abrogated MK polarization might result in an insufficient and nondirectional proplatelet formation because Pdk1−/− MKs fail to contact the sinusoids, essential for sufficient intrasinusoidal sequestration of platelets, and displayed a significant reduction of platelet production in vitro and in vivo. Besides defective actin polymerization on collagen I–coated surfaces, Pdk1-null MKs displayed a diminished spreading area as well as a reduced number and abnormal morphology of podosomes. MK podosomes consist of an F-actin–rich core and appear to be important for MKs to extend proplatelet protrusions through the basement membrane of the blood vessel and platelet delivery into the bloodstream during later stages of platelet formation.28,43
Genetic knockout of several regulatory proteins of the MK cytoskeleton, such as the GTPases RhoA9,44 and Cdc4212 or the kinase PAK2,29 has been associated with pronounced macrothrombocytopenia with only moderately increased platelet turnover, but defective proplatelet formation and significantly increased number of MKs in the BM of the mutant mice, comparable to the present findings in Pdk1−/− mice. F-actin–dependent formation of the DMS in maturating MKs requires the activation of Cdc42 and its effector PAK, an axis facilitating the partitioning of internal membranes and nuclei into polarized territories.8 Recently, it has been shown that the small GTPases Cdc42 and RhoA act as a regulatory circuit to coordinate MK polarization in transendothelial platelet biogenesis and that this process might involve PI3K-dependent signaling.9 Interestingly, although Pdk1−/− MKs displayed a substantial defect in polarization and proplatelet formation, activity of the GTPases Cdc42 and RhoA, both regulators of PAK signaling as well, was not significantly affected in Pdk1-deficient MKs (supplemental Figure 9). These data indicate that PDK1 may regulate MK actin cytoskeleton dynamics and polarization, both essential for sufficient platelet production, either downstream or independently from signaling involving GTPases RhoA or Cdc42.
As reported by Dütting et al, pharmacological PI3K inhibition reduced the number of MKs in direct contact with sinusoids, but had no effect on DMS development or total MK numbers in the BM.9 Similar to our findings after MK-specific deletion of Pdk1, Pak2 deficiency in MKs resulted in an impaired actin cytoskeleton assembly with an altered DMS ultrastructure, augmented MK endomitosis with consecutively upregulated MK polyploidization, and increased extramedullary thrombopoiesis due to impaired thrombopoiesis in the BM.29 Besides its ability to selectively bind Rho GTPase Cdc42, resulting in an increase of its activity, PAK is sufficiently activated by PDK1 via phosphorylation.30 In fact, we found an abrogated phosphorylation of PAK (Thr423) in Pdk1−/− MKs. Of note, evidence for PI3K-independent PDK1-dependent phosphorylation of PAK has been reported earlier.30 In a next step, we could show that the phosphorylation-dependent activation of LIMK and consecutive inactivation of cofilin (by phosphorylation at Ser3), which both are essential downstream effectors of PAK, were similarly abrogated in Pdk1-deficient MKs. In a nonphosphorylated state, cofilin stimulates severing and depolymerization of actin.45 Thus, comparable to previous findings in dendritic cells,19 increased cofilin activity in Pdk1−/− MKs due to reduced cofilin phosphorylation suggested that the slower rate of F-actin recovery in Pdk1-null MKs may result from an increased depolymerization activity of cofilin in these cells. In line with our findings in Pdk1-deficient MKs, enhanced actin severing and depolymerization in MKs have previously been associated with increased MK polyploidization.46,47 MKs with genetic deficiency in Rac1/Cdc42 displayed increased phosphorylation of LIMK and cofilin and no difference in MK ploidy, suggesting that PDK1-dependent signaling via PAK in the LIMK/cofilin-dependent regulation of MK actin cytoskeleton and polyploidization does not involve GTPases Rac1/Cdc42.
Another member of the AGC family of protein kinases that is regulated by PDK1 and potentially involved in the development of the MK actin cytoskeleton and the process of MK polyploidization is the Rho-associated protein kinase ROCK. As reported in cancer cells, PDK1 controls cortical actinomyosin contraction and myosin light chain phosphorylation via activation of ROCK1 that functions downstream of the GTPase RhoA.20 In MKs, inhibition of RhoA/ROCK pathway led to decreased F-actin at the cleaved furrow with increased ploidy,48,49 a phenotype comparable to MKs with Pdk1 deficiency. Furthermore, genetic deletion of RhoA resulted in increased MK ploidy, impaired MK polarization, and significant macrothrombocytopenia.9,44,50 However, RhoA activity was not significantly affected in Pdk1−/− MKs (supplemental Figure 9). Further studies will be necessary to address the question of whether PDK1 may influence ROCK-dependent processes in megakaryopoiesis independently from RhoA.
Besides PAK, atypical PKCζ has been implicated as direct substrate for phosphorylation by PDK1.51 PKCζ was identified as an essential regulator of cellular polarization in neurons,52 and PDK1-dependent signaling via atypical PKC is essential for cellular polarity in epithelia.53,54 Consistent with these previous findings in other cell types, we could show that PKCζ phosphorylation was significantly reduced in Pdk1-deficient MKs. Moreover, these results indicate that impaired activation of atypical PKC isoform PKCζ might be involved in the defective MK polarization of Pdk1−/− MKs. Nevertheless, further studies will be necessary to elucidate the exact molecular mechanisms.
In conclusion, using MK/platelet–specific deletion of PDK1, the present study discloses a key role for the protein kinase PDK1 in the tightly regulated signaling processes orchestrating MK cytoskeleton organization, polyploidization, and proplatelet formation during thrombopoiesis. PDK1 is critically required for MK F-actin cytoskeleton dynamics during maturation, podosome formation, and DMS polarization. PDK1-dependent signaling in MKs during thrombopoiesis involves the PAK/LIMK/cofilin phosphorylation cascade as well as atypical PKCζ. These findings may deepen the understanding of (patho-)physiological mechanisms underlying proplatelet formation, thrombopoiesis, and the development of macrothrombocytopenia.
All datasets and protocols may be found in the supplemental material and methods available with the online version of this article. For further information, please contact the corresponding author.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank Daniela Eißler for excellent technical assistance and gratefully acknowledge the generosity of Dario R. Alessi, MRC Protein Phosphorylation Unit, University of Dundee, UK, who provided the Pdk1fl/f mice. They thank the microscopy platform of the Bioimaging Centre (Rudolf Virchow Centre) for providing technical infrastructure and support.
This study was funded by the Deutsche Forschungsgemeinschaft (German Research Foundation), project number 374031971-TR 240. The authors acknowledge support by the Open Access Publishing Fund of the University of Tübingen.
The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Authorship
Contribution: S.G., I.P., H.S., and O.B. designed the research; S.G., K.A., I.P., G.M., C.B., M.-C.M., P.M., B.W.-A., D.S., M.M., C.E.B., and L.Q.-F. conducted the experiments; S.G., K.A.,. D.S., G.M., D.R., T.G., H.R.S., P.S., F.L., B.N., M.G., H.S., I.P., and O.B. analyzed the data; S.G. and O.B. wrote the manuscript; and K.A., C.E.B., F.L., B.N., M.G., H.S., and I.P. contributed to the writing of the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Oliver Borst, Department of Cardiology, Angiology & Cardiovascular Medicine, University of Tübingen, Otfried Müller-Str 10, 72076 Tübingen, Germany; e-mail: oliver.borst@med.uni-tuebingen.de.

![MK-specific PDK1 deficiency results in macrothrombocytopenia and a reduced platelet lifespan. (A) Arithmetic means ± standard error of the mean (SEM; n = 6, unpaired 2-tailed Student t test) of Pdk1 transcript levels in platelets and kidneys from Pdk1fl/fl and Pdk1−/− mice. Representative immunoblot of PDK1 protein levels in MKs from Pdk1fl/fl and Pdk1−/− mice (n = 5). mRNA, messenger RNA. (B) Arithmetic means ± SEM of platelet count and mean platelet volume of Pdk1fl/fl and Pdk1−/− mice aged 8 weeks (n = 13), 12 weeks (n = 22), and 18 weeks (n = 10). (C) Arithmetic means ± SEM (n = 12) of mean forward scatter (FSC) signal of Pdk1fl/fl and Pdk1−/− platelets. (D) Arithmetic means ± SEM (n = 8, unpaired 2-tailed Student t test) of relative spleen weight in Pdk1fl/fl and Pdk1−/− mice. (E) TPO levels of Pdk1fl/fl and Pdk1−/− mice. Arithmetic means ± SEM (n = 6, unpaired 2-tailed Student t test) are shown. TPO receptor/c-Mpl protein level in Pdk1fl/fl and Pdk1−/− MKs. Actin was used as loading control. Arithmetic means ± SEM (n = 5, unpaired 2-tailed Student t test) are shown. (F) Platelet counts of Pdk1fl/fl and Pdk1−/− mice before and 4 days after treatment with TPO (2 µg per animal per day). Arithmetic means ± SEM (n = 8, 2-way analysis of variance [ANOVA] with Bonferroni correction for multiple comparisons) are shown. (G) Endogenous survival of Pdk1fl/fl and Pdk1−/− platelets measured by determination of percent fluorescently labeled platelets in vivo at indicated time points after injection of DyLight 488 α-glycoprotein IX (GPIX) (n = 6, unpaired 2-tailed Student t test). (H) Platelet depletion in Pdk1fl/fl and Pdk1−/− mice was induced by IV injection of rat anti-GPIbα antibody (Emfret Analytics). Peripheral platelet counts were determined over a time course of 7 days by flow cytometry. The rate of platelet count recovery as compared with the respective initial platelet count is depicted (n = 4, unpaired 2-tailed Student t test). *P < .05 and **P < .01 as well as ##P < .01 indicate statistically significant differences.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/134/21/10.1182_blood.2019000185/7/m_bloodbld2019000185f1.png?Expires=1766979532&Signature=sIIiQP90dtlcux-oYX2vSMATdPsgQLjo5cmeU-9af7PgaQHdA6ArhAYsJE5EUsH602RTvjRWDPGXCiVNxlWEU7ISNS1Tor6eD8VbT2QuO87SCgA8vNNmpA-YxNslrFouN4rfqDpquLnmnZHblKSR99-PCwQopn9EUKl171qs2qAiCpBdMAEhnGhbYqKq5TGpo4Bvk5Od03JwZYaG7xqUJxr57lFhoFNKD~z8tyl9oxXZC0EheaQAq0JyWPSrZZO9jsZCX8LHyJOS6POdO6DV~7sSK0wh-x7gUddIyiuwYn9yKP3Xs1sQltaQq7mJ-utP6Zd6BwOVshQods5ruuhlCg__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
![Pdk1−/−mice display increased localization of MKs in the BM hematopoietic compartment with less sinusoidal contact, a substantial BM MK hyperplasia, and an upregulation of extramedullary thrombopoiesis. (A) Representative images of GPIb immunostaining of BM sections from 2-month-old Pdk1fl/fl and Pdk1−/− mice (left). Quantification of Pdk1fl/fl and Pdk1−/− BM MKs per visual field. Arithmetic means ± SEM (n = 6, unpaired 2-tailed Student t test; right) are shown. Scale bar equals 50 μm. (B) Representative images of hematoxylin and eosin stainings as well as GPIb immunostaining of spleen sections from 6-month-old Pdk1fl/fl and Pdk1−/− mice (left). Quantification of Pdk1fl/fl and Pdk1−/− spleen MKs per visual field. Arithmetic means ± SEM (n = 6, unpaired 2-tailed Student t test; right) are shown. Scale bar equals 50 μm. (C) Representative confocal microscopy images of immunostained BM sections (n = 5). Green, MKs (GPIb); red, sinusoids (endoglin, CD105); gray, nuclei (4′,6-diamidino-2-phenylindole [DAPI]). Scale bar equals 20 μm. Boxes in the left panels mark the section in the illustrations which are highlighted in a higher magnification in the right panels. (D) Arithmetic means ± SEM (n = 6, unpaired 2-tailed Student t test) of MKs per visual field in immunostained BM sections of Pdk1fl/fl and Pdk1−/− mice. (E) Quantification of MK distance from sinusoids in BM sections of Pdk1fl/fl and Pdk1−/− mice (means ± SEM, n = 6, 750 MKs, Wilcoxon-Mann-Whitney test). (F) Quantification of MK localization in the BM of Pdk1fl/fl and Pdk1−/− mice (arithmetic means ± SEM, n = 6, 2-way ANOVA with Bonferroni correction for multiple comparisons). BMHC, bone marrow hematopoietic compartment; Intrasin., intrasinusoidal; SC, sinusoidal contact. **P < .01 indicates statistically significant difference.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/134/21/10.1182_blood.2019000185/7/m_bloodbld2019000185f2.png?Expires=1766979532&Signature=J-n9uh5ltewfgX2ocW15BmJe1hTDtGaNyhtnKWp8WR-7zfIRH~eRAifbNhk3-zzzsgBXrGXkf5uAboi48JqygD2Q1sWg3fu2S0NFHuZscJ05m8hzhibzPr6e7dBnTw4Q9KjHbJE1C518NkGTKuNJmXhvk10STnhkuJAi7wX-xVuhUbmxC-8E6MA1W4rup8bjhC9Yro-Ukn2U3TN2fS-rOxpO6vnk2UGzXMWnt5qi1zVfUOlKOKb1kzdmKwzz8A4CWJvALzrXgkdNtCdQ1xfIK7glYPfWtdc~kEAXNMA-~vjavyrI9c1PuLrozQWucJIbw~4c7Da-YllSJ25sYY6lBw__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)