Key Points
Direct anticoagulants are currently used in patients with thromboembolism, irrespective of the presence of antiphospholipid antibodies.
This trial shows an increased rate of events with rivaroxaban compared with warfarin in patients with antiphospholipid syndrome.
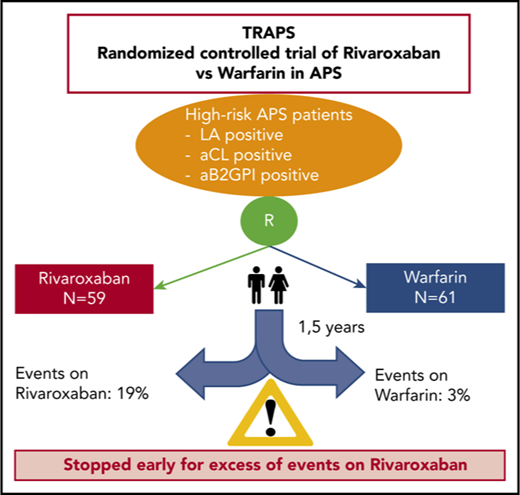
Rivaroxaban is an effective and safe alternative to warfarin in patients with atrial fibrillation and venous thromboembolism. We tested the efficacy and safety of rivaroxaban compared with warfarin in high-risk patients with thrombotic antiphospholipid syndrome. This is a randomized open-label multicenter noninferiority study with blinded end point adjudication. Rivaroxaban, 20 mg once daily (15 mg once daily based on kidney function) was compared with warfarin (international normalized ratio target 2.5) for the prevention of thromboembolic events, major bleeding, and vascular death in patients with antiphospholipid syndrome. Only high-risk patients triple positive for lupus anticoagulant, anti-cardiolipin, and anti–β2-glycoprotein I antibodies of the same isotype (triple positivity) were included in the study. The trial was terminated prematurely after the enrollment of 120 patients (59 randomized to rivaroxaban and 61 to warfarin) because of an excess of events among patients in the rivaroxaban arm. Mean follow-up was 569 days. There were 11 (19%) events in the rivaroxaban group, and 2 (3%) events in the warfarin group. Thromboembolic events occurred in 7 (12%) patients randomized to rivaroxaban (4 ischemic stroke and 3 myocardial infarction), whereas no event was recorded in those randomized to warfarin. Major bleeding occurred in 6 patients: 4 (7%) in the rivaroxaban group and 2 (3%) in the warfarin group. No death was reported. The use of rivaroxaban in high-risk patients with antiphospholipid syndrome was associated with an increased rate of events compared with warfarin, thus showing no benefit and excess risk. This trial was registered at www.clinicaltrials.gov as #NCT02157272.

Introduction
Antiphospholipid syndrome (APS) is an acquired autoimmune disease that is characterized by the association of thromboembolic events or pregnancy morbidity and the presence of antiphospholipid (aPL) antibodies.1 The laboratory tests exploring the presence of aPL antibodies include lupus anticoagulant (LA), anti-cardiolipin (aCL) antibodies, and anti–β2-glycoprotein I (aβ2GPI) antibodies. Laboratory tests may be positive in different combinations; however, a particular group of individuals, those presenting with positivity for all 3 laboratory tests (triple positive), are at the highest risk for both a first thrombotic event and a higher rate of recurrence despite antithrombotic treatment.2,,,,-7 Triple-positive individuals make up <50% of those positive for 1 or 2 tests exploring the presence of aPL antibodies.4 Secondary prevention of thromboembolic events is the primary therapeutic goal in these patients, and the mainstay treatment is warfarin, because it significantly reduces thromboembolic recurrences.8,-10
However, the management of warfarin therapy is challenging, and thromboembolic events are frequent when the intensity of anticoagulation is not adequate (ie, International Normalized Ratio [INR] <2).11 Major bleeding is also a concern among APS patients when INR is kept at target values >3.11,12 An anticoagulant with a predictable effect and no need for monitoring would be of interest to the young population of patients with thrombotic APS. Rivaroxaban, a direct inhibitor of factor Xa, is at least as effective as warfarin in preventing venous13 and arterial thromboembolism.14,15 Moreover, it exhibits a safer profile due to a significantly lower incidence of cerebral bleeding.14 Under these premises, we designed and conducted a randomized controlled trial aiming to compare the efficacy and safety of rivaroxaban and warfarin in APS patients (Trial on Rivaroxaban in AntiPhospholipid Syndrome [TRAPS]) at high-risk for thromboembolic recurrence.
Methods
Study design
TRAPS is a prospective randomized phase 3 open-label noninferiority study with blinded end point adjudication conducted in 14 centers in Italy. The study design has been described previously.16 The protocol was approved by the Institutional Review Board at each participating site. An independent Data and Safety Monitoring Board whose members were unaware of treatment allocation closely monitored the trial.
Patients and randomization
Adult patients were eligible for inclusion if they were between the ages of 18 and 75 years, were positive for all 3 aPL tests in the last blood sampling (triple positivity), and had a history of thrombosis (objectively proven arterial, venous, and/or biopsy-proven microthrombosis). Triple positivity was defined as positivity for immunoglobulin G (IgG) and/or IgM aCL (>40 GPL [G phospholipid units] or MPL [M phospholipid units] or greater than the 99th percentile), IgG, and/or IgM aβ2GPI (>40 U or greater than the 99th percentile) and LA test based on the recommendations of the International Society of Thrombosis and Hemostasis.17,18 aCL and aβ2GPI ELISA tests had to be positive for the same isotype. At the screening visit, as well as at follow-up visits, recent laboratory tests (complete blood count, renal and liver function, and standard coagulation tests) were examined. After signing an informed consent form, patients underwent Web-based randomization using random block sizes of 2, 4, and 6 and stratified based on sex and the presence or absence of an associated autoimmune disease. In this way, 4 strata were constructed: females with or without associated autoimmune disease and males with or without associated autoimmune disease. Exclusion criteria are detailed elsewhere.16 Randomization and study data were managed using Research Electronic Data Capture19 tools hosted at the Department of Cardiac Thoracic and Vascular Sciences, Padua University Hospital.
Study drug regimen
Patients randomized to rivaroxaban received 20 mg once daily if they had a creatinine clearance (CrCl) >50 mL/min, as determined by the Cockroft–Gault method. In case of CrCl between 30 and 50 mL/min, 15 mg of rivaroxaban was administered once daily. Patient compliance was checked by counting residual pills at each follow-up visit. Patients exiting the study were not considered in compliance calculation. Patients on warfarin randomized to rivaroxaban started treatment when INR was <3. If the patient was naive to anticoagulation and randomized to warfarin, patient maintenance dose was determined using the protocol endorsed by the Italian Federation of Anticoagulation Clinics.20 INR was maintained between 2.0 and 3.0 and checked at least every 4 weeks, or more frequently if needed, at the investigator’s discretion.
Follow-up and outcomes
Enrolled patients underwent regularly scheduled visits 1 and 3 months after randomization and every 6 months thereafter. At each visit, compliance to rivaroxaban was checked by residual pill count, and compliance to warfarin was checked by calculating the time in therapeutic range. Temporary drug discontinuation for surgery or invasive procedures was carried out according to established protocols.21,22 The reasons for final premature drug discontinuation were recorded. The use of aspirin in addition to anticoagulant drugs was allowed at the investigator’s discretion.
The primary outcome was the cumulative incidence of thromboembolic events, major bleeding, and vascular death. Diagnosis of venous and arterial thromboembolism was based on objective imaging techniques, diagnosis of major bleeding was according to published guidelines,23 and diagnosis of vascular death was ascertained from clinical or autopsy reports and/or death certificates.16
Statistical analysis
The primary analysis was designed to test whether the experimental drug (rivaroxaban) is noninferior to the active control (warfarin). The noninferiority margin was set to correspond to the preservation of 50% of the warfarin effect, as adopted in the other rivaroxaban registration trials. The noninferiority margin is derived from the only observational study comparing vitamin K antagonists with control in triple-positive APS patients.4 This study showed a rate of composite events (thrombosis, major bleeding, and death) among APS patients receiving warfarin of ∼6% per year. On the other hand, the incidence of events in untreated patients is 13% per year. The hazard ratio (HR) margin corresponding to preservation of 50% of the warfarin effect is 1.7. To satisfy the noninferiority hypothesis, the upper boundary of the 1-sided 95% confidence interval (CI) for the HR of an outcome with rivaroxaban compared with warfarin must fall below 1.7. A noninferiority log-rank test with 88 overall events and sample size of 536 subjects (268 in the reference group and 268 in the treatment group) achieves 80% power at a 0.05 1-tail significance level to detect an equivalence HR of 1.7.
The primary outcome was analyzed according to “as treated” and intention to treat (ITT) principle; “as treated” analysis included patients who completed the study on the assigned treatment on randomization, whereas ITT included all patients who underwent randomization. Final analysis included events that occurred from the randomization date to 25 January 2018 (the day on which investigators were instructed to switch patients to a nonstudy vitamin K antagonist). Baseline continuous and categorical data are reported as appropriate: means and standard deviations for continuous data and as numbers and percentages for categorical data. Cox proportional-hazards modeling was used for the end point analyses. Statistical significance was set at P ≤ .05.
Results
Trial enrollment began on 2 November 2014 and was stopped ahead of the planned date on 25 January 2018 by the Advisory Board, based on recommendations of the Adjudication and Safety Committee. All participating patients discontinued the assigned study drug and switched to a nonstudy vitamin K antagonist. At the time of trial termination, 120 patients had been randomized: 59 in the rivaroxaban group and 61 in the warfarin group. Patient characteristics are described in Table 1. Randomization produced a satisfactory balance between groups for the considered variables. Risk factors for thrombosis were also comparable between the 2 study groups. Two patients met the dose-reduction criteria based on calculated CrCl and received 15 mg of rivaroxaban.
There were 9 (12%) patients in the rivaroxaban group and 3 (5%) in the warfarin group who permanently stopped their assigned therapy before an end point event and before the termination date. Reasons for discontinuation are shown in Table 2. The mean follow-up period was 569 days for the “as treated” cohort and 611 days for the ITT cohort. No patient was lost to follow-up. Adherence to treatment was 96% in the rivaroxaban group, and the time in therapeutic range was 67% in the warfarin group.
Clinical outcomes
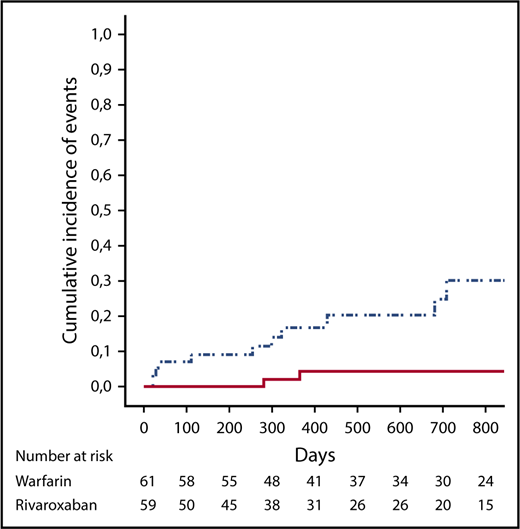
Overall, 13 events were reported during follow-up in the “as treated” population (Table 3). A post hoc ITT analysis included 2 additional events. In the “as treated” analysis (Table 4; Figure 1), the composite primary outcome of thromboembolic events, major bleeding, and vascular death occurred in 11 patients in the rivaroxaban group and in 2 patients in the warfarin group (HR, 6.7; 95% CI, 1.5-30.5; P = .01). In the rivaroxaban arm, ischemic stroke occurred in 4 (7%) patients, and myocardial infarction occurred in 3 (5%) patients, whereas there were no cases of ischemic stroke or myocardial infarction in the warfarin group. Of the 7 patients experiencing arterial events in the rivaroxaban arm, 1 patient was on aspirin; conversely, among the 52 patients without arterial events, 10 patients were on aspirin (P = not significant). No episode of venous thromboembolism was recorded in either arm. There were 4 and 2 cases of major bleeding in the rivaroxaban and warfarin groups, respectively (HR, 2.5; 95% CI; 0.5-13.6; P = .3). Overall, bleeding events were associated with predisposing factors in 5 of 6 cases (uterine fibroma, anal fissure, and Crohn’s disease, as well as thrombocytopenia in 2 patients). None of the patients treated with the lower-dose rivaroxaban had an event. No patient died during the follow-up.
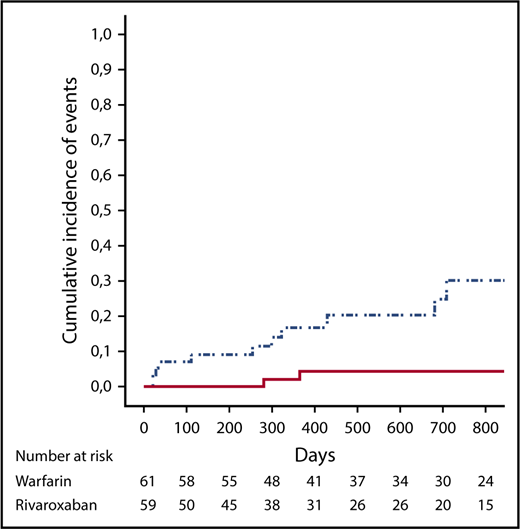
Cumulative incidence of events (death, thromboembolic events, and major bleeding) in the rivaroxaban group (dotted and dashed line) and in the warfarin group (solid line).
Cumulative incidence of events (death, thromboembolic events, and major bleeding) in the rivaroxaban group (dotted and dashed line) and in the warfarin group (solid line).
In the ITT analysis (Table 4) that includes the 12 patients who exited the study prematurely, the composite primary outcome of thromboembolic events, major bleeding, and vascular death occurred in 13 patients in the rivaroxaban group and in 2 patients in the warfarin group (HR, 7.4; 95% CI, 1.7-32.9; P = .008). There were 2 additional events in the rivaroxaban arm with respect to the “as treated” analysis. One bilateral deep vein thrombosis of the lower limbs was recorded 21 days from stopping the study drug (rivaroxaban) for gingival bleeding while the patient was treated with therapeutic doses of low molecular weight heparin. One cardiovascular death occurred 433 days from study drug (rivaroxaban) suspension in a patient with a history of congestive heart failure and while the patient was on warfarin.
Discussion
The aim of the present trial was to evaluate whether a direct oral anticoagulant, rivaroxaban, was noninferior to warfarin in terms of efficacy and safety in high-risk patients with thrombotic APS. However, the trial was stopped prematurely for an excess of events in the rivaroxaban arm. Thromboembolic events, all in the arterial circulation, primarily drove the imbalance in the cumulative primary end point. Four ischemic strokes and 3 myocardial infarctions occurred in patients treated with rivaroxaban, whereas none occurred in patients taking warfarin. Thus, rivaroxaban apparently does not protect high-risk patients from arterial events. Because there is no strict concordance between the arterial and venous sites of the qualifying event at diagnosis and that of the recurrent thrombotic event,4 it does not mean that rivaroxaban can be safely used in APS patients with venous thromboembolism at diagnosis. Three of 7 cases with an arterial outcome were originally patients with venous thromboembolism. Reasons for rivaroxaban failure remain elusive. Possible explanations may be related to poor adherence,24 insufficient drug concentration, or a different mechanism of action with respect to warfarin. In our study, the excellent adherence, based on pill count, does not explain treatment failure in the rivaroxaban arm. A suboptimal drug concentration may account for thromboembolic complications during treatment.25 The requirement for higher anti-Xa activity and plasma rivaroxaban levels for the prevention of arterial vs venous events has been demonstrated in animal models. Preclinical data showed an effective dose in 50% of animals (ED50) of 0.1 mg/kg vs 5.0 mg/kg for the prevention of venous (venous stasis model) versus arterial (arteriovenous shunt model) thrombosis in rats.26 Although pharmacological studies have demonstrated a predictable rivaroxaban anticoagulant effect, high interindividual variability may expose some patients to inadequate plasma levels of the drug.27 Differences in the mechanisms of action of rivaroxaban and warfarin may also, in part, explain our findings. Thrombin generation in APS patients treated with rivaroxaban is different compared with warfarin, because vitamin K antagonists reduce functional coagulation factors in the extrinsic and intrinsic pathways of coagulation.28 The importance of the intrinsic pathway in thrombin generation is highlighted by the ability of warfarin to better attenuate thrombin generation with prosthetic material.29
Platelets may play a major role in thrombus formation in the arterial circulation, as shown in an animal model of aPL-induced arterial thrombosis.30 Whether additional therapy with aspirin is efficacious in APS patients is not known. To this end, conclusions cannot be drawn from this trial, because only a small proportion of patients were taking aspirin, and they were well balanced in the 2 groups. Moreover, there was no statistical difference between patients who had arterial events on rivaroxaban and were treated with aspirin and patients on rivaroxaban and not receiving aspirin who did not experience arterial events. Hydroxychloroquine was suggested to play a role in lowering aPL antibody titers and preventing thrombotic recurrences in APS.31,32 In our study, the small proportions of hydroxychloroquine-treated patients were balanced in the 2 arms, and a clear correlation of its use with events cannot be established.
Previous reports and systematic reviews were inconclusive regarding the benefit or harm of direct oral anticoagulants in APS.33,34 Major biases in the reported studies were the heterogeneous aPL profiles and the lack of reports on drug compliance.33 A previous randomized trial of rivaroxaban vs warfarin (Rivaroxaban in Antiphospholipid Syndrome [RAPS]) with a biological end point was conducted in APS patients with venous thromboembolism.28 However, there are differences between the TRAPS and RAPS trials: the uniform high-risk patient population (triple positivity) enrolled in TRAPS, the inclusion of APS patients with both arterial and venous qualifying events, and the clinical end point. A strength of the TRAPS trial is that it was designed to confirm clinical efficacy and long-term safety of rivaroxaban in thrombotic APS; although the trial was not completed, results strongly suggest the lack of benefit of its use in APS patients.
The results of our trial should raise awareness among health care personnel about the lack of efficacy of rivaroxaban in high-risk triple-positive APS patients, and they may be relevant for the other trials testing direct anticoagulants in this setting.35,36 In fact, the Apixaban for the Secondary Prevention of Thromboembolism Among Patients with the AntiphosPholipid Syndrome (ASTRO-APS) investigators had to modify their protocol as a result of an increased incidence of arterial events.37 Results from this study cannot be translated to APS patients without a “full-positive” laboratory profile. At present, the therapeutic strategy in patients with a laboratory profile different from that of this study should be considered on a case-by-case basis, taking into account the presence of additional risk factors for venous and arterial thrombosis,38 the nature of venous thromboembolism (provoked or unprovoked), and the risk for bleeding.
In conclusion, rivaroxaban in high-risk patients with APS was associated with an excess of events compared with warfarin.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
M. Taraborelli (University of Brescia), E. Silvestri (University of Florence), T. Del Ross (University of Padua), and A. Doria (University of Padua) contributed to patient enrollment.
This work was supported by funding from Bayer S.p.A. (V.P.)
Authorship
Contribution: V.P. conceived and planned the study and took the lead in writing the manuscript; G.Z., S.P.J., A.H., A.R., L.A., A. Tincani, C.C., D.P., T.F., P.G., A.C., V.D.M., A.G., A.F., I.M., S.T., D.B., and M.G. made substantial contributions to acquisition and interpretation of data; G.D. and A. Tosetto computed data and performed data analysis; A.B. monitored the database; and all authors critically revised the manuscript and gave their approval to the final version.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Vittorio Pengo, Clinical Cardiology, Thrombosis Center, Department of Cardiac Thoracic and Vascular Sciences, Via Giustiniani 2, 35128 Padua, Italy; e-mail vittorio.pengo@unipd.it.
Appendix
Study coordinator: V. Pengo; Steering Committee: V. Pengo, A.R., A. Tincani, and P. L. Meroni (University of Milan); Independent Blinded Safety and Event-Adjudication Committee: G. Palareti (University of Bologna) and P. Prandoni (University of Padua).