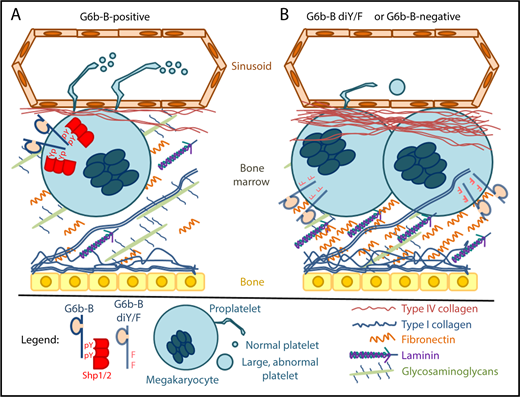
In both humans and mice, megakaryocytes that express a functional form of G6b-B (A) reside in a bone marrow environment that contains normal levels of extracellular matrix proteins. Phosphorylation of tyrosine residues (pY) within G6b-B ITIM and ITSM motifs enables recruitment and activation of Shp1 and/or Shp2, which are required for production of normal numbers of properly functioning platelets. Megakaryocytes that do not express G6b-B or that express a dysfunctional form of G6b-B that cannot be tyrosine phosphorylated (G6b-B di-Y/F; B) form clusters in a fibrotic bone marrow environment and produce low numbers of large, dysfunctional platelets. These findings demonstrate that the signaling capabilities of G6b-B play an important role in regulating platelet number and function, and identify pathogenic variants in the gene that encodes G6b-B as a cause of inherited myelofibrosis. Figure design was inspired by Leiva et al.13
In both humans and mice, megakaryocytes that express a functional form of G6b-B (A) reside in a bone marrow environment that contains normal levels of extracellular matrix proteins. Phosphorylation of tyrosine residues (pY) within G6b-B ITIM and ITSM motifs enables recruitment and activation of Shp1 and/or Shp2, which are required for production of normal numbers of properly functioning platelets. Megakaryocytes that do not express G6b-B or that express a dysfunctional form of G6b-B that cannot be tyrosine phosphorylated (G6b-B di-Y/F; B) form clusters in a fibrotic bone marrow environment and produce low numbers of large, dysfunctional platelets. These findings demonstrate that the signaling capabilities of G6b-B play an important role in regulating platelet number and function, and identify pathogenic variants in the gene that encodes G6b-B as a cause of inherited myelofibrosis. Figure design was inspired by Leiva et al.13
G6b-B, which is encoded by a gene that is variously called G6b, C6orf25, or MPIG6B, is a platelet- and megakaryocyte-specific receptor that binds the tandem Src homology 2 domain-containing protein tyrosine phosphatases Shp2 and Shp1 upon phosphorylation of cytoplasmic immunoreceptor tyrosine-based inhibitory and switch motifs (ITIM and ITSM, respectively).3,,,-7 Mice lacking G6b-B,8 or whose platelets lack Shp1 and Shp2,9 exhibit severe macrothrombocytopenia, defects in proplatelet formation, and aberrant platelet function.
Hofmann et al validate the relevance of findings made in G6b-B–deficient mice to human disease by demonstrating the association, in humans, of germ line loss-of-function mutations in G6b with congenital macrothrombocytopenia with focal myelofibrosis (cMFTM), which is a rare form of childhood myelofibrosis. Furthermore, they demonstrate that G6b-B–deficient mice also exhibit hematologic and histopathologic features of myelofibrosis, which can be rescued by expression of human G6b-B. Together with the recent report of thrombocytopenia in a family with a truncation mutation in G6b,10 these results strongly support the conclusion that G6b-B is required in both mice and humans for normal production of platelets by megakaryocytes in the bone marrow.
Geer et al use mice engineered to express a mutant form of G6b-B in which the ITIM and ITSM tyrosine residues were substituted with phenylalanine (G6b-B diY/F) to address the mechanism by which G6b-B functions in megakaryocytes and platelets. Specifically, they demonstrate that G6b-B diY/F mice phenocopy the severe macrothrombocytopenia, defective proplatelet production, aberrant platelet function, and myelofibrosis that are characteristic of G6b-B–deficient mice and humans. These results indicate that G6b-B requires its ITIM and ITSM, presumably to become phosphorylated and bind Shp1 and Shp2, to support normal platelet production by megakaryocytes (see figure).
The association of G6b-B with inherited myelofibrosis has implications for future studies. First, these studies justify additional clinical research to determine the extent to which loss-of-function mutations in G6b contribute to primary myelofibrosis in children, the causes of which are notably distinct from the identically named adult disease and remain largely uncharacterized.11 Second, more basic scientific work is needed to identify the ligands with which G6b-B normally interacts in the bone marrow, the kinases that are responsible for its phosphorylation, and how it works with Shp1/2 to support normal hematopoiesis and (pro)platelet production.12
Conflict-of-interest disclosure: The author declares no competing financial interests.