

Abstract
Mastocytosis is a rare, clinically heterogenous clonal hematological neoplasm. Over 95% of patients harbor the driver KIT D816V mutation resulting in mast cell (MC) accumulation and proliferation in various organs, leading to variable symptom manifestations that result from MC mediator release in patients with systemic mastocytosis (SM) and end-organ damage in those with advanced SM. The accurate diagnostic and clinical classification of patients with SM is vital to underpin appropriate treatment options and personalize therapy. This review evaluates the current diagnostic criteria, clinical classification, risk stratification, and therapeutic options available for adult patients with nonadvanced and advanced SM.
Learning Objectives
Review the diagnostic criteria and classification of mastocytosis
Summarize the current treatment options for patients with SM
Discuss the impact of potent TKIs in the current and future management landscape for patients with SM
CLINICAL CASE
A 45-year-old man with a 20-year history of biopsy- proven KIT D816V mutation-positive urticaria pigmentosa developed flushing and gastrointestinal symptoms. He subsequently had a life-threatening anaphylactic episode precipitated by aspirin, requiring intensive care. The following year he was treated for an acute gastrointestinal hemorrhage due to severe gastritis and multiple gastro-duodenal erosions. CT imaging showed enlarged retroperitoneal and mediastinal lymph nodes and hepatosplenomegaly. Serum tryptase was 525 µg/L and alkaline phosphatase 315 IU/L. A bone marrow biopsy revealed 50% round and spindle-shaped mast cells (MCs) with no evidence of associated myeloid neoplasm (AMN). Immunohistochemistry showed strong expression for MC tryptase, CD117, CD25, CD2, and CD30. G-banding chromosomal analysis was normal, and next-generation sequencing did not detect any additional myeloid mutations. His enlarging spleen with progressive lymphadenopathy and raised alkaline phosphatase (ALP) (>2.5 × ULN) were considered “C-findings” clinically to classify him as having aggressive systemic mastocytosis, and he was started on midostaurin 100 mg twice daily with an initial good clinical response. After 18 months, serum tryptase plateaued at approximately 250 µg/L with no change in the MC burden in the bone marrow. He was enrolled in the PATHFINDER study and received avapritinib 200 mg daily. There was resolution of palpable hepatomegaly and splenomegaly, and the serum tryptase had normalized to 11 µg/L within 2 months. Repeat bone marrow assessment showed no clonal MCs with normalization of all C findings and blood counts, indicating he had achieved a complete remission. Avapritinib was reduced to 100 mg daily and at his 2.5-year follow-up, he remains in a complete remission.
This case illustrates a patient who would have been diagnosed with MC in the skin and then confirmed as smoldering systemic mastocytosis (SSM) when he had a bone marrow biopsy with high MC disease burden (MC disease burden 50% and tryptase >200 µg/L) and mild organomegaly when he was referred and developed significant mediator-related symptoms. He progressed to aggressive systemic mastocytosis (SM) (organomegaly and evidence of end-organ damage) 2 decades after his initial presentation to the dermatologist and responded to first- and second-line tyrosine kinase inhibitor (TKI) therapy. Most patients with indolent systemic mastocytosis, however, do not progress to advanced disease, and those patients with high disease burden such as those with smoldering mastocytosis should be monitored closely.
Figure 1 montage summarizes the case with a descriptive legend.
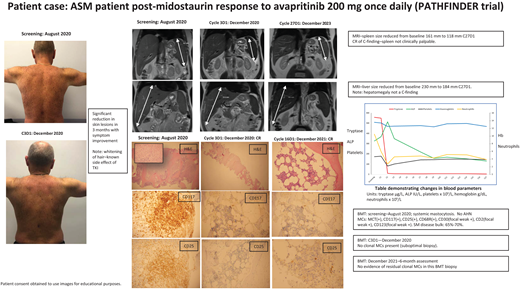
Clinical case summary of ASM patient's response to second-line TKI treatment. In patients with pure MCL and ASM, treatment with midostaurin can result in a PR as our case illustrates. This patient had a diagnosis of c-KIT–positive ASM with AMN nor any additional myeloid mutations. He was initially treated with midostaurin 100 mg twice a day. His symptoms and MC disease burden improved, as reflected by reduced spleen size, improvement of skin rash, and decreased tryptase levels. After 18 months his symptoms recurred, including fatigue, sweats, worsening rash, and increased splenomegaly and tryptase levels. He had lost his response to midostaurin. He subsequently was enrolled in the PATHFINDER trial and received avapritinib 200 mg once daily initially. As seen in Figure 1, he had an excellent response, achieving a CR within 3 months of treatment, which has been maintained. He experienced the expected side effects of mild periorbital edema, whitening of his hair, and an “ALP flare,” which is seen with initiation of TKI treatment and normalizes in 4 to 6 weeks. As reported in PATHFINDER, patients with pure ASM achieve a deep, fast response despite previous exposure to another TKI—midostaurin, in his case. Should he have an allogenic bone marrow transplant? The current view is that because he has no AMN and only harbors the c-KIT mutation, he should continue on a reduced dose of avapritinib 100 mg once daily. It will be interesting to see if this dose could be reduced or interrupted to see if he maintains a CR in the future. BMT, bone marrow trephine; H&E, hematoxylin and eosin stain; MRI, magnetic resonance imaging.
Clinical case summary of ASM patient's response to second-line TKI treatment. In patients with pure MCL and ASM, treatment with midostaurin can result in a PR as our case illustrates. This patient had a diagnosis of c-KIT–positive ASM with AMN nor any additional myeloid mutations. He was initially treated with midostaurin 100 mg twice a day. His symptoms and MC disease burden improved, as reflected by reduced spleen size, improvement of skin rash, and decreased tryptase levels. After 18 months his symptoms recurred, including fatigue, sweats, worsening rash, and increased splenomegaly and tryptase levels. He had lost his response to midostaurin. He subsequently was enrolled in the PATHFINDER trial and received avapritinib 200 mg once daily initially. As seen in Figure 1, he had an excellent response, achieving a CR within 3 months of treatment, which has been maintained. He experienced the expected side effects of mild periorbital edema, whitening of his hair, and an “ALP flare,” which is seen with initiation of TKI treatment and normalizes in 4 to 6 weeks. As reported in PATHFINDER, patients with pure ASM achieve a deep, fast response despite previous exposure to another TKI—midostaurin, in his case. Should he have an allogenic bone marrow transplant? The current view is that because he has no AMN and only harbors the c-KIT mutation, he should continue on a reduced dose of avapritinib 100 mg once daily. It will be interesting to see if this dose could be reduced or interrupted to see if he maintains a CR in the future. BMT, bone marrow trephine; H&E, hematoxylin and eosin stain; MRI, magnetic resonance imaging.
Introduction
Mastocytosis is a rare clonal MC disease, driven by a somatic mutation in the KIT gene (D816V) in more than 90% of adults with the disease, resulting in the expansion and accumulation of neoplastic MCs in various tissues, including skin, bone marrow, gastrointestinal tract, liver, and/or spleen.1,2 The symptoms that can affect mastocytosis patients3 are shown in Figure 2.
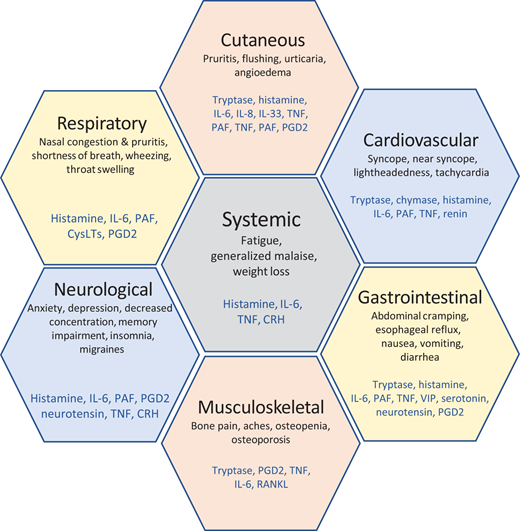
MC mediator release with biological and clinical consequences. CRH, corticotropin-releasing hormone; CysLTs, cysteinyl leukotrienes; IL-6/8/33, interleukin 6/8/33; PAF, platelet-activating factor; PGD2, prostaglandin D2; RANKL, receptor activator of nuclear factor kβ ligand; TNF, tumor necrosis factor; VIP, vasoactive intestinal peptide. Adapted from Theoharides et al.3
MC mediator release with biological and clinical consequences. CRH, corticotropin-releasing hormone; CysLTs, cysteinyl leukotrienes; IL-6/8/33, interleukin 6/8/33; PAF, platelet-activating factor; PGD2, prostaglandin D2; RANKL, receptor activator of nuclear factor kβ ligand; TNF, tumor necrosis factor; VIP, vasoactive intestinal peptide. Adapted from Theoharides et al.3
Each patient is unique in how mastocytosis affects them. This can present diagnostic challenges as patients may initially present to several clinicians before a formal diagnosis is made.4
Mastocytosis is classified into 3 subtypes: cutaneous mastocytosis (CM), SM, and MC sarcoma (MCS). CM is characterized by atypical MCs limited to the skin only and is more common in children than adults. SM is characterized by the infiltration of clonal MCs in the bone marrow and other extracutaneous organs. Diagnosis of SM is usually confirmed by a bone marrow biopsy. There are 2 subcategories of SM: nonadvanced SM and advanced SM (AdvSM).1,2,5 Up to 80% of SM patients have indolent disease with a variable symptom burden but a normal life expectancy. Patients with AdvSM have a poor prognosis as a result of MC infiltration leading to end-organ damage with overall survival (OS) ranging from 2 to 6 months for patients with MC leukemia (MCL) and 2 to 4 years for patients with SM and an associated myeloid neoplasm (SM-AMN).6,7
Classification
The World Health Organization (WHO) 2022 classification of myeloid neoplasms2 recognizes 6 SM subtypes: bone marrow mastocytosis (BMM), indolent SM (ISM), smoldering SM (SSM), aggressive SM (ASM), MC leukemia (MCL), and SM with an associated hematologic neoplasm (SM-AHN). BMM, ISM, and SMM are regarded as nonadvanced while ASM, MCL, and SM-AHN are regarded as advanced SM. BMM was added as a new SM subtype characterized by the absence of skin lesions and B-findings and a serum tryptase below 125 µg/L, whereas the ICC1 regards it as a subtype of ISM.
The International Consensus Classification 2022 (ICC) classifies ISM and SSM in the nonadvanced SM category, reflecting that there is no end-organ damage in normal hematological and biochemical parameters and no significant organomegaly.
ICC has modified the subtype of SM-AHN to SM with an associated myeloid neoplasm (SM-AMN) to reflect that the associated neoplasm typically shares a KIT mutation and/or other clonal genetic abnormalities is typically myeloid. To diagnosis SM-AMN, the patient must meet the diagnostic criteria for SM and for an associated myeloid neoplasm (e.g. chronic myelomonocytic leukaemia or other myelodysplastic/myeloproliferative neoplasms, myelodysplasia, myeloproliferative neoplasms, acute myeloid leukaemia, or other myeloid neoplasm), and the associated myeloid neoplasm should be fully classified according to established criteria (Table 1).
ICC and WHO 2022 classification for the diagnosis of mastocytosis
| WHO | ICC | |
|---|---|---|
| CM Urticaria pigmentosa/maculopapular cutaneous mastocytosis • Monomorphic variant • Polymorphic variant Diffuse cutaneous mastocytosis Cutaneous mastocytoma • Isolated mastocytoma • Multilocalized mastocytoma | CM Urticaria pigmentosa/maculopapular cutaneous mastocytosis Diffuse cutaneous mastocytosis Mastocytoma of skin | |
| SM BMM* ISM SSM ASM SM-AHN MCL MC sarcoma | SM ISM (includes BMM) SSM ASM SM-AMN MCL MC sarcoma | |
| B-findings | >30% BM cellularity by MCs in histology and serum tryptase >200 ng/ml and/or KIT D816V VAF >10% in BM or PB leukocytes | >30% of BM cellularity by MC aggregates (assessed on BM biopsy) and serum tryptase >200 ng/mL |
| Hypercellular BM with loss of fat cells and prominent myelopoiesis ± left shift and eosinophilia ± leukocytosis and eosinophilia and/or discrete signs of myelodysplasia (<10% neutrophils, erythrocytes, and megakaryocytes) | Cytopenia (not meeting criteria for C-findings) or cytosis. Reactive causes are excluded, and criteria for other myeloid neoplasms are not met. | |
| Palpable hepatomegaly without ascites or other signs of organ damage and/or palpable splenomegaly without hypersplenism and without weight loss and/or lymphadenopathy (>2 cm) | Hepatomegaly without impairment of liver function and/or splenomegaly without features of hypersplenism including thrombocytopenia and/or lymphadenopathy (>1 cm size) on palpation or imaging | |
| BMM: no B-findings, absence of skin lesionsa basal serum tryptase <125 mg ISM <2 B-findings SSM ≥2 B-findings | ISM <2 B-findings SSM ≥2 B-findings | |
| C-findings | Cytopenias: ANC <1 × 109/L and/or Hb <100 g/L and/or platelets <100 × 109/L | |
| Hepatopathy: ascites and elevated liver enzymes ± hepatomegaly or cirrhotic liver ± portal hypertension | ||
| Spleen: palpable splenomegaly with hypersplenism ± weight loss ± hypoalbuminemia | ||
| GI tract: malabsorption with hypoalbuminemia ± weight loss | ||
| Bone: large-sized osteolysis (≥2 cm) with pathologic fracture bone pain ASM 1 or more C-findings/SM-AHM/AMN and MCL may not have C-findings | ||
| WHO | ICC | |
|---|---|---|
| CM Urticaria pigmentosa/maculopapular cutaneous mastocytosis • Monomorphic variant • Polymorphic variant Diffuse cutaneous mastocytosis Cutaneous mastocytoma • Isolated mastocytoma • Multilocalized mastocytoma | CM Urticaria pigmentosa/maculopapular cutaneous mastocytosis Diffuse cutaneous mastocytosis Mastocytoma of skin | |
| SM BMM* ISM SSM ASM SM-AHN MCL MC sarcoma | SM ISM (includes BMM) SSM ASM SM-AMN MCL MC sarcoma | |
| B-findings | >30% BM cellularity by MCs in histology and serum tryptase >200 ng/ml and/or KIT D816V VAF >10% in BM or PB leukocytes | >30% of BM cellularity by MC aggregates (assessed on BM biopsy) and serum tryptase >200 ng/mL |
| Hypercellular BM with loss of fat cells and prominent myelopoiesis ± left shift and eosinophilia ± leukocytosis and eosinophilia and/or discrete signs of myelodysplasia (<10% neutrophils, erythrocytes, and megakaryocytes) | Cytopenia (not meeting criteria for C-findings) or cytosis. Reactive causes are excluded, and criteria for other myeloid neoplasms are not met. | |
| Palpable hepatomegaly without ascites or other signs of organ damage and/or palpable splenomegaly without hypersplenism and without weight loss and/or lymphadenopathy (>2 cm) | Hepatomegaly without impairment of liver function and/or splenomegaly without features of hypersplenism including thrombocytopenia and/or lymphadenopathy (>1 cm size) on palpation or imaging | |
| BMM: no B-findings, absence of skin lesionsa basal serum tryptase <125 mg ISM <2 B-findings SSM ≥2 B-findings | ISM <2 B-findings SSM ≥2 B-findings | |
| C-findings | Cytopenias: ANC <1 × 109/L and/or Hb <100 g/L and/or platelets <100 × 109/L | |
| Hepatopathy: ascites and elevated liver enzymes ± hepatomegaly or cirrhotic liver ± portal hypertension | ||
| Spleen: palpable splenomegaly with hypersplenism ± weight loss ± hypoalbuminemia | ||
| GI tract: malabsorption with hypoalbuminemia ± weight loss | ||
| Bone: large-sized osteolysis (≥2 cm) with pathologic fracture bone pain ASM 1 or more C-findings/SM-AHM/AMN and MCL may not have C-findings | ||
BM, bone marrow; PB, peripheral blood.
B-findings reflect high MC disease burden, and C-findings indicate organ damage caused by MC infiltration and are used to distinguish between ISM, SMM, and ASM. Some minor refinements have been made in the ICC, namely the presence of cytopenias not meeting the criteria for C-findings. In the WHO classification, KIT D816V mutation with a variant allele frequency (VAF) >10% now qualifies as a B-finding. Highly sensitive polymerase chain reaction (PCR) assays, eg, digital droplet PCR or allele-specific PCR, should be used to detect and quantify the KIT VAF. Next-generation sequencing to identify the presence of high-risk mutations SRSF2, ASXL1, and RUNX1 (S/A/R panel) enables prognostic risk stratification and presence of myeloid mutations, eg, TET2, JAK2, DNMT3A, NRAS,CBL, and EZHZ, which may be detected in patients who have an AHN/AMN.
The current 2 histological classifications, WHO and ICC, are not aligned and therefore can cause confusion.
Diagnostic criteria
The ICC and WHO have introduced some refinements to the diagnostic criteria for SM (Table 2). Demonstration of tryptase and KIT (CD117) positivity by immunohistochemistry have been added to enable proper identification of MCs in stained sections. The presence of CD30 and of any activating KIT mutation have been added as minor diagnostic criteria. An adjustment method for tryptase levels has been proposed for patients proven to have hereditary α tryptasemia.
ICC and WHO 22 diagnostic criteria for the diagnosis of SM
| ICC Presence of the major criterion is sufficient for diagnosis. In the absence of the major criterion, at least 3 of the 4 minor criteria must be present. |
|---|
| Major criterion Multifocal dense infiltrates of tryptase- and/or CD117-positive MCs (≥15 MCs in aggregates) detected in sections of BM and/or other extracutaneous organ(s) |
| Minor criteria a. In BM biopsy or in section of other extracutaneous organs >25% of MCs are spindle-shaped or have an atypical immature morphology b. MCs in BM, PB, or other extracutaneous organs express CD25, CD2, and/or CD30, in addition to MC markers c. KIT D816V mutation or other activating KIT mutation detected in BM, PB, or other extracutaneous organs d. Elevated serum tryptase level, persistently >20 ng/mL In cases of SM-AMN, an elevated tryptase does not count as an SM minor criterion. |
| WHO Requires at least 1 major criterion and 1 minor or 3 minor criteria. |
| Major criterion Multifocal dense infiltrates of MCs (≥15 MCs in aggregates) in BM biopsies and/or in sections of other extracutaneous organ(s) |
| Minor criteria a. >25% of all MCs are atypical cells (type I or type II) on BM smears or are spindle-shaped in MC infiltrates detected on sections of visceral organs b. KIT point mutation at codon 816 in the BM or another extracutaneous organ c. MCs in BM, blood, or another extracutaneous organ exhibit CD2 and/or CD25 d. Baseline serum tryptase level >20 ng/mL In case of an unrelated myeloid neoplasm, item d is not valid as an SM criterion. |
| ICC Presence of the major criterion is sufficient for diagnosis. In the absence of the major criterion, at least 3 of the 4 minor criteria must be present. |
|---|
| Major criterion Multifocal dense infiltrates of tryptase- and/or CD117-positive MCs (≥15 MCs in aggregates) detected in sections of BM and/or other extracutaneous organ(s) |
| Minor criteria a. In BM biopsy or in section of other extracutaneous organs >25% of MCs are spindle-shaped or have an atypical immature morphology b. MCs in BM, PB, or other extracutaneous organs express CD25, CD2, and/or CD30, in addition to MC markers c. KIT D816V mutation or other activating KIT mutation detected in BM, PB, or other extracutaneous organs d. Elevated serum tryptase level, persistently >20 ng/mL In cases of SM-AMN, an elevated tryptase does not count as an SM minor criterion. |
| WHO Requires at least 1 major criterion and 1 minor or 3 minor criteria. |
| Major criterion Multifocal dense infiltrates of MCs (≥15 MCs in aggregates) in BM biopsies and/or in sections of other extracutaneous organ(s) |
| Minor criteria a. >25% of all MCs are atypical cells (type I or type II) on BM smears or are spindle-shaped in MC infiltrates detected on sections of visceral organs b. KIT point mutation at codon 816 in the BM or another extracutaneous organ c. MCs in BM, blood, or another extracutaneous organ exhibit CD2 and/or CD25 d. Baseline serum tryptase level >20 ng/mL In case of an unrelated myeloid neoplasm, item d is not valid as an SM criterion. |
BM, bone marrow; PB, peripheral blood.
Risk stratification
Schwab et al. demonstrated that 89% of patients with AdvSM harbored additional somatic aberrations, the most commonly affected genes being TET2, SRSF2, ASXL1, RUNX1, and CBL.8 Mutated genes in the S/A/R panel are associated with inferior OS in SM patients and are considered high-risk mutations. Jawar et al. analyzed 70 SM patients and demonstrated a clear difference in 3-year OS between patients with 0 mutations (90% OS), 1 mutation (73% OS), and ≥2 mutations (42% OS) in the S/A/R gene panel.9,10 Patients with additional somatic mutations detected were in the SM-AMN group, and associated myeloid neoplasm was noted.
Prognostic scoring systems integrating clinical and molecular characteristics in patients with SM have evolved: the International Prognostic Scoring System for Mastocytosis,7 the Mayo Alliance Prognostic System,11 the Mutation-Adjusted Risk Score,12 and the Global Prognostic Score for Mastocytosis.13 The validated Mutation-Adjusted Risk Score scoring model for patients with AdvSM (n = 383) identified from multivariate analysis that inferior OS was associated with age >60 years, anemia (Hb <100 g/L), thrombocytopenia (platelet count <100 × 109/L), and the presence of 1 or ≥2 high-risk mutations (S/A/R panel). Low-risk, intermediate-risk, and high-risk groups were devised based on these parameters, with respective OS times not reached, 4.3 years and 1.9 years.
Well-differentiated systemic mastocytosis
In the rare (<5%) separate morphological variant of SM known as well-differentiated systemic mastocytosis, MCs are usually round, hypergranulated, and lacking CD25 and CD2 expression but frequently expressing CD30. Mutations in extracellular, juxtamembrane, and transmembrane domains in exons 8 through 10 may be seen, eg, K509I and F522C, which are imatinib sensitive, while KIT is usually wild type with KIT D816V or exon 17 mutations not detected.14
Approach to diagnosis
Diagnosis requires a multidisciplinary, integrated approach. Ideally, patients should be referred to a specialist center. Patients with a suspected diagnosis of SM need a comprehensive symptom review and clinical examination. Most adult patients will have CM. A minority of patients present with osteoporosis, idiopathic anaphylaxis, or significant mediator symptoms lacking skin lesions and may have BMM. The presence of lymphadenopathy/organomegaly points to a possible diagnosis of AdvSM. Patients with idiopathic anaphylaxis and BMM may have tryptase levels lower than 20 µg/L, and applying the REMA score or the European Competence Network on Mastocytosis/Fuchs score may be helpful when considering whether a bone marrow biopsy is warranted.15
Baseline blood tests include a full blood count with differential as the latter may subtly indicate the presence of an AMN, eg, monocytosis/eosinophilia. A blood film is informative in AdvSM cases; morphological signs of dysplasia, atypical monocytes suggestive of a myelodysplastic or myelomonocytic component, leukoerythroblastic blood film, leukocytosis, or thrombocytosis may point to a myeloproliferative disorder or myeloid leukemia. Circulating MCs confer a diagnosis of MCL; in acute MCL these are medium/large atypical blastic MCs with coarse metachromatic granules with pleomorphic nuclei and nucleoli. In chronic MCL they are round, hypergranular forms with inconspicuous nuclei. Serum tryptase, liver, and renal profiles and lactate dehydrogenase are useful. Bone marrow biopsy can confirm SM diagnosis. In clinical practice it is vital to review any histology looking for an occult associated myeloid-neoplasm, if somatic mutations in addition to KIT D816V are detected. The reverse applies when patients with the most commonly associated myeloid neoplasm, CMML, also have a KIT D816V mutation detected, then clinician should look for clonal mast cells.
A DEXA scan is required in all adult patients at diagnosis and on follow-up for assessment of T scores to evaluate for osteoporosis or osteopenia. Generally patients with ISM osteoporosis need treating while osteosclerosis is an expected finding in patients with AdvSM. The need for additional radiological investigation, such as skeletal survey for lytic lesions or magnetic resonance imaging for sclerotic lesions, should be guided by clinical presentation.
Treatment options
Nonadvanced SM
The mainstay of nonadvanced SM management in most patients is symptom control with a combination of antimediator medications16 (Figure 3). Identified triggers exacerbating symptoms should be avoided. Antihistamine medications (both anti H1 and anti H2)17,18 in combination with MC-stabilizing agents are effective, the latter with gastrointestinal symptoms.19,20 A short course of corticosteroids helps minimize “flares” of severe symptoms. In a small cohort of patients, leukotriene receptor inhibitors (Montelukast) or cyclo-oxygenase inhibitors (Aspirin) may be of use.21,22 Neuropathic symptoms may be managed with tricyclic antidepressants and anticonvulsants (eg, gabapentin). Medications will need adjustment due to fluctuating patient symptoms, with higher doses required than recommended in national formularies. Immune tolerance therapy is recommended for patients with BMM or ISM presenting with anaphylaxis due to bee/wasp venom allergies.23,24 Omalizumab has shown benefit in this subset of patients.25-27 Some patients experience significant symptoms despite combinations of high doses of antimediator treatments and have a poor quality of life (QoL). These refractory patients should be considered for cytoreductive therapy with cladribine (2CdA)28,29 or targeted TKI agents if available. Avapritinib, elenastinib (BLU-263/HARBOR), masitinib, and bezuclastinib (CGT9486/SUMMIT) are currently being evaluated within trials.
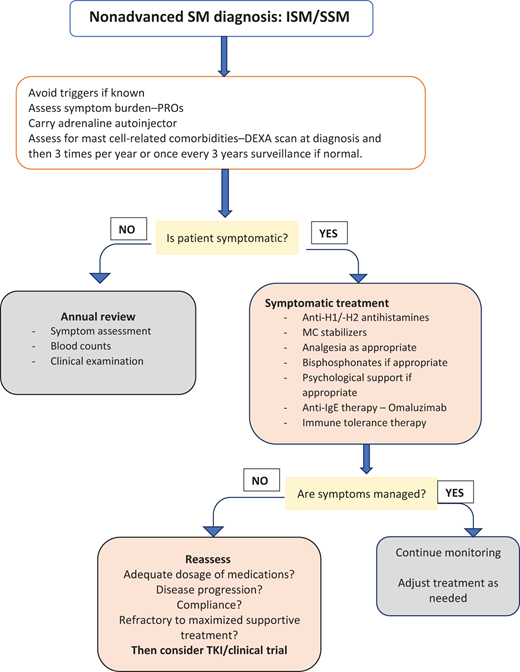
Management of nonadvanced SM. PROs, patient-reported outcomes. Adapted from Gerds AT, Gotlib J, Ali H, et al. Systemic mastocytosis, version 1.2022, https://www.nccn.org/guidelines. Clinical Practice in Oncology (NCCN Guidelines): National Comprehensive Cancer Network; 2022.
Management of nonadvanced SM. PROs, patient-reported outcomes. Adapted from Gerds AT, Gotlib J, Ali H, et al. Systemic mastocytosis, version 1.2022, https://www.nccn.org/guidelines. Clinical Practice in Oncology (NCCN Guidelines): National Comprehensive Cancer Network; 2022.
In May 2023 avapritinib was approved by the Food and Drug Administration for the treatment of symptomatic ISM patients upon meeting the primary end points in the PIONEER trial (Blueprint Medicines Corporation; ClinicalTrials.gov number NCT03731260).30 This double-blind, placebo-controlled, phase 2 trial randomized patients with moderate to severe ISM (total symptom score [TSS] ≥28) 2:1 to avapritinib 25 mg once daily (n = 141) or placebo (n = 71), both with BSC. The primary end point was mean change in TSS (range 0-110) based on the 14-day average of patient-reported severity of 11 symptoms. Secondary end points included ≥50% and ≥30% reduction in TSS; ≥50% reductions in serum tryptase, blood KIT D816V VAF, and bone marrow MCs; and QoL measures. Primary and key secondary end points were assessed from baseline to week 24. Avapritinib significantly improved TSS (mean change −15.6 vs −9.2; P = 0.003) and the likelihood of achieving a ≥50% TSS (25% vs 10%; P = 0.005) and ≥30% TSS reduction (45% vs 30%; P = 0.009) compared with placebo. A greater proportion of avapritinib-treated patients achieved ≥50% reductions in serum tryptase, KIT D816V VAF, and bone marrow MC burden (all P < 0.0001). QoL scores showed up to 4.1-fold greater improvement with avapritinib versus placebo. Safety profiles were similar between treatment groups with few discontinuations due to adverse events (AEs). Avapritinib 25 mg once daily appeared to be well tolerated by most patients.
Advanced systemic mastocytosis
Over the last decade, TKIs have moved to the forefront of treatment in the limited licensed therapeutic options available for AdvSM patients. Patients usually presenting with end-organ damage due to MC infiltration should be considered for cytoreductive or targeted treatment with TKIs (Figure 4).
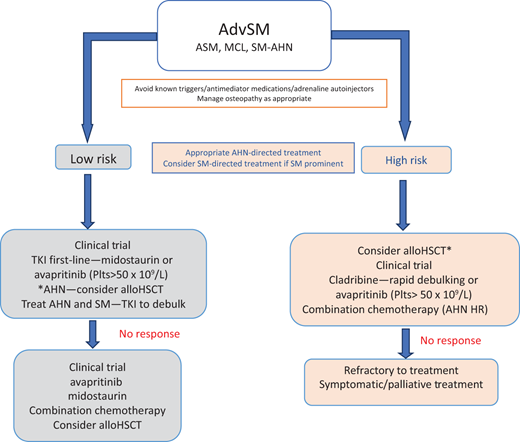
Management options for AdvSM. HR, high risk; Plts, platelets. *If AHN is the major component contributing to patient symptoms. Reproduced with permission from Radia DH, Moonim MT. Update on diagnostic approaches and therapeutic strategies in systemic mastocytosis. Best Pract Res Clin Haematol. 2022;35(2):101380. doi:10.1016/j.beha.2022.101380.
Management options for AdvSM. HR, high risk; Plts, platelets. *If AHN is the major component contributing to patient symptoms. Reproduced with permission from Radia DH, Moonim MT. Update on diagnostic approaches and therapeutic strategies in systemic mastocytosis. Best Pract Res Clin Haematol. 2022;35(2):101380. doi:10.1016/j.beha.2022.101380.
Treatment options
TKIs
Imatinib. Twose et al. reported on the efficacy of imatinib in KIT D816V–negative well-differentiated systemic mastocytosis patients treated with 300 or 400 mg daily for 12 months31,32 with an overall response rate (ORR) of 50%. Imatinib is also effective in patients with SM and a coexisting chronic eosinophilic leukemia associated with the PDGFR-α/β rearrangements. The lack of activity against the common KIT D816V mutation excludes it as a treatment in most SM patients. Imatinib was approved in 2006 for the treatment of adult ASM patients with wild-type KIT or unknown mutational status.
Midostaurin has in vitro activity against kinase domain KIT D816V and D816Y mutations and FMS-related tyrosine kinase 3, PDGRF α/β, and vascular endothelial growth receptor 2 mutations. Midostaurin gained approval in 2016 following results from a multicenter international phase 2, single-arm study of 116 patients demonstrating a favorable safety and efficacy profile.33 Patients received midostaurin 100 mg twice daily until disease progression or unacceptable toxicity. Results from 89 patients in the primary efficacy population of the study (16 ASM; 16 MCL; 57 SM-AHN) with a median follow-up of 26 months (range 12-54 months) demonstrated an ORR of 60% (45% major response and 15% partial response) as per the modified Valent and modified Cheson criteria. The median duration of response was 24.1 months, median OS 28.7 months, and median progression-free survival (PFS) 14.1 months. Response rates reported were ASM, 75%; SM-AHN, 58%; and MCL 50%, regardless of prior therapy, AHN status, or KIT D816V status. Significant decreases were seen in tryptase levels and bone marrow MC burden in >50% and spleen size in 77% of patients with improvements in C-findings. Objective improvements in disease-related symptoms and skin lesions were noted. Eighty-two percent of patients reported mild gastrointestinal AEs at all grades, with 6% to 8% experiencing grade 3-4 symptoms. Nausea and vomiting (related to the capsule) were the main adverse symptoms, and most patients tolerated midostaurin with adjunctive antiemetic medication (ondansetron + domperidone). Myelosuppression seen in patients with cytopenias (neutropenia [24%], anemia [41%], and thrombocytopenia [29%]) was managed with dose reduction and growth factor support.
Additional analysis of 38 patients treated with midostaurin (in the global trial or the compassionate-use program) reported poorer outcomes and survival of patients who harbored additional high-risk mutations (S/A/R) and did not achieve a >25% reduction in their KIT VAF.34 The development of additional mutations and an increase in the VAF of non–KIT D816V mutations (K/N -RAS, RUNXI, IDH2, and NPM1) was seen in these patients with disease. No acquired resistance mutations were noted on midostaurin treatment.
Avapritinib is a potent and selective oral type 1 multi- kinase inhibitor with activity against KIT D816V, targeting active kinase formation and preventing it from binding to its substrates. Avapritinib was approved in patients with AdvSM with a platelet count >50 × 109/L by the Food and Drug Administration in June 2021 and European Medicines Agency in March 2022 following data reported from the registrational PATHFINDER study. This single-arm, phase 2 study demonstrated the efficacy and safety of avapritinib with a starting dose of 200 mg once daily in patients with AdvSM, excluding patients with SM-AML and high-risk MDS.35 A prespecified interim analysis was carried out in 32 response-evaluable patients within the PATHFINDER study using modified IWG-MRT-ECNM criteria. The median follow-up was 10.4 months with a confirmed ORR (complete remission [CR], CR with partial hematologic recovery [CRh], partial remission [PR], and CR with incomplete count recovery [CI]) of 75% (n = 24). A modified response criteria CRh was developed for patients who achieved a complete pathological response to the SM component (CR demonstrated in the bone marrow with loss of MC aggregates, normalization of tryptase, and spleen size) but had partial hematological recovery (hemoglobin of >80 but <100 g/L, platelet counts between 50-100 × 109/L, and neutrophil count between 0.5-1.0 × 109/L). The myelosuppressive effect of avapritinib and/or the presence of a concomitant AMN may be responsible for the partial hematological recovery. A CRh was reported in 6 patients (19%), PR in 10 patients (31%), and CI in 8 patients (25%). Responses were seen regardless of prior therapy and presence of mutations in the S/A/R panel. Significant reductions in tryptase, bone marrow MC burden, and KIT D816V VAF of at least 50% from the baseline were seen in 93%, 88% and 60% of the patients, respectively. The most frequently reported AEs were peripheral and periorbital oedema (50% and 48%), mainly at grade 1-2 and managed with diuretics and dose reductions. Grade 3 neutropenia, thrombocytopenia, and anemia was seen in 24%, 16%, and 16% of patients, respectively. Gastrointestinal AEs were predominately grade 1-2 with diarrhea (23%), nausea (18%), and vomiting (18%). Cognitive impairment presenting as mild memory impairment was reported in 6 patients (grade 1-2). One patient had a subdural hematoma prior to protocol amendment, which excluded patients with a platelet count of <50 × 109/L. No treatment-related deaths occurred.
Most patients' C-findings improved from baseline. At the time of data cutoff, the median PFS and median OS in the safety population (n = 62) had not been reached. The estimated 12-month PFS and OS rates were 79% and 86%, respectively with 52 patients (84%) still on treatment, with a median follow-up of 7 months (range 5.6-8.1 months).
In a subanalysis from the phase 1 EXPLORER study, which looked at the mutational landscape in 69 patients, with a median follow-up of 23 months, 20% (14 patients) progressed on treatment, with progression driven by KIT D816V–negative AMN clones in most cases.36,37 In these patients the KIT D816V VAF remained low, suggesting that treatment directed to the AMN component was appropriate.
There are no head-to-head comparisons in trials of avapritinib to midostaurin or cladribine. A global multicenter retrospective review of patient charts was carried out in 6 study sites in the US, Europe, and the UK collecting data from AdvSM patients who received best available therapy (BAT) in routine clinical practice using inclusion and exclusion criteria similar to those for the EXPLORER and PATHFINDER trials.38 The study population included 176 avapritinib patients and 141 BAT patients, contributing to 222 lines of treatment. Patients treated with BAT contributed data on multiple lines of therapy, and these data were compared with pooled patient data from both trials. In the BAT arm, 50% of the patients had been exposed to midostaurin and 25% to cladribine. Results showed longer treatment (avapritinib 30.6 months vs BAT 5.5 months), a greater reduction in tryptase level (avapritinib 86.6% vs BAT 9.2%), and significantly longer OS (inverse probability treatment weighting–adjusted median OS was avapritinib 49 months vs BAT 26.8 months). This real-world study showed the improved efficacy of avapritinib compared with other available therapies for AdvSM used in clinical practice.
Bezuclastinib is an oral highly selective TKI with potent activity against KIT D816V. It has demonstrated preliminary clinical activity and a tolerable safety profile in a phase 1/2 study of patients with advanced solid tumors, including gastrointestinal stromal tumor. A reduction in KIT exon 17 mutational burden was noted in patients treated with bezuclastinib.38 Bezuclastinib has been designed to avoid activity against other related kinases, eg, PDGFRα, PDGFRβ, wild-type KIT, VEGFR2 (KDR), and CSF1R (FMS), in order to avoid related AEs. APEX, a multicenter, phase 2, open-label, 2-part clinical study to evaluate the safety, efficacy, pharmacokinetics, and pharmacodynamics of bezuclastinib in subjects with AdvSM, is currently recruiting patients, with promising preliminary data.39
Cytoreductive therapy
Cladribine is a cytoreductive agent delivered at a dose of 0.14 mg/kg intravenous or subcutaneous infusions over 5 days, repeated every 4 to 12 weeks. Barete et al. reported 32 patients with AdvSM treated with cladribine; the median number of courses was 3.7 (range 1 to 9) with a 50% ORR (major remission/ PR) and median duration of response of 2.47 years (range 0.5-8.6 years) in this cohort. Myelosuppression led to the most common grade 3/4 serious AEs due to lymphopenia (82%), neutropenia (47%), and opportunistic infections (13%).29 In current clinical practice cladribine is considered in patients with rapidly progressive AdvSM where fast debulking of disease is required to stabilize the patient and potentially introduce targeted TKI treatment, or in those who cannot be treated with TKI.28,29
SM-AHN
The current treatment algorithm for patients with SM-AMN directs treatment to each of the 2 components as if the other were not present. Because most studies suggest that AMN progression is likely, the AMN component is treated invariably first. In some patients with ISM-AHN (eg, ISM- CMML1), an option may be to monitor until one or the other component progresses. It is not always easy to ascertain which of the 2 neoplasms is contributing to a patient's clinical symptoms and organopathy. The patient's mediator symptom profile with diagnostic tests (FBC, ALP, bone marrow biopsy, KIT VAF, and next-generation sequencing for myeloid mutations) will provide an overall picture to inform management decisions (Figure 5).
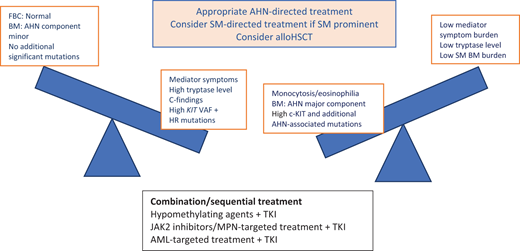
Considerations in the treatment of patients with SM-AHN. BM, bone marrow; FBC, full blood count; HR, high risk; MPN, myeloproliferative neoplasms. Reproduced with permission from Radia DH, Moonim MT. Update on diagnostic approaches and therapeutic strategies in systemic mastocytosis. Best Pract Res Clin Haematol. 2022;35(2):101380. doi:10.1016/j.beha.2022.101380.
Considerations in the treatment of patients with SM-AHN. BM, bone marrow; FBC, full blood count; HR, high risk; MPN, myeloproliferative neoplasms. Reproduced with permission from Radia DH, Moonim MT. Update on diagnostic approaches and therapeutic strategies in systemic mastocytosis. Best Pract Res Clin Haematol. 2022;35(2):101380. doi:10.1016/j.beha.2022.101380.
Allogeneic hematopoietic stem cell transplantation
Ustin et al. reported on retrospective outcome data in 201440 from a large multicenter cohort of 57 patients with SM who had an allogeneic bone marrow transplant (38 SM-AHN, 7 ASM, and 12 MCL). The response rate was 70%, with a 16% CR. Of the 30% of patients who did not have a response, 21% had stable disease and 9% had primary refractory disease. Although CR was achieved in all 38 patients with SM-AHN within the AHN component, 10 patients progressed with a relapse of the AHN, and 50% of these patients did not survive. Median 3-year OS for the cohort was 57% (74% SM-AHN, 43% ASM, and 17% MCL). Treatment-related mortality at 6 months and 1 year was 11% and 20%, respectively, and was highest for MCL patients. A diagnosis of MCL and use of reduced-intensity allogeneic transplant was associated with poor OS. Parameters affecting OS or PFS were patient and donor age, donor type, graft source, KIT mutation status, karyotype, and conditioning with or without total-body irradiation. The challenge with allogeneic hematopoietic stem cell transplant (alloHSCT) is for patients to have minimal disease, ideally CR in both SM and AMN components to optimize outcomes. The new potent TKIs, eg, avapritinib, that may result in SM CR/CRh moves alloHSCT up the treatment algorithm for eligible patients.40-42
Conclusion
Continued education is necessary for health care professionals and patients to recognize the heterogenous presentation of patients with mastocytosis and therefore reduce the time to a formal diagnosis of systemic mastocytosis. Unfortunately, the disharmony of complex diagnostic and classification systems leads to confusion in classifying patients. Health care professionals must work together internationally to ensure that this is not detrimental to patient care and outcomes.
TKIs have heralded a new era in the management of patients with SM. Midostaurin paved the way in demonstrating disease modification that leads to MR/PR with improvement in QoL. Avapritinib has demonstrated improved QoL with a reduction in symptom burdens in patients with debilitating ISM and significant improvement in OS and QoL in AdvSM patients. This enables eligible patients to be considered for a curative alloHSCT, although several questions need to be addressed to optimize outcomes—eg, timing of transplant, when to stop TKI, and monitoring of SM components pre- and posttransplant. The exciting next step is investigating the impact of sequential or combined treatment for patients with SM-AMN through international collaborative trials. Further research to evaluate new diagnostic markers and therapeutic strategies (eg, CD30, PD L1) is ongoing. The European Competence Network on Mastocytosis and the American Initiative in Mast Cell Diseases have proposed a new set of response criteria which include pathologic, molecular, cytogenetic, clinical, and symptom/QoL response within a 4-tiered schema.43 Validation of better clinical scoring systems for both prognosis and response assessments are exciting avenues to continue improving outcomes for mastocytosis patients.
Acknowledgments
The authors thank the MPNSM team at Guy’s & St Thomas' NHS Foundation Trust and members of the European Competence Network on Mastocytosis with American Initiative in Mast Cell Diseases for spearheading international collaborative research in this rare group of disorders.
Conflict-of-interest disclosure
Scott Veitch: no competing financial interests to declare.
Deepti H. Radia: Clinical advisory board/study steering group member (EXPLORER, PATHFINDER, AZURE); research support and educational events/Speaker for Blueprint Medicines Corporation. Consultant and Study Steering Group member (APEX) Cogent Biosciences educational events/speaker: Novartis. Royalties: Fast Facts Systemic Mastocytosis coauthor, Karger.
Off-label drug use
Scott Veitch: Nothing to disclose.
Deepti H. Radia: Nothing to disclose.
