
Abstract
This chapter reviews several important themes pertaining to the antiphospholipid syndrome (APS), including a description of the clinical features, a discussion of the main autoantigen, beta 2-glycoprotein I (β2GPI), and insights into the characteristics of the pathogenic anti-β2GPI autoantibodies. Evidence-based considerations for when to test for APS are explored, along with the clinical significance of patients testing positive on multiple APS assays, so-called triple positivity. A detailed review of recently published laboratory guidelines for the detection of lupus anticoagulant and the solid-phase anticardiolipin and anti-β2GPI ELISAs is undertaken. Finally, a brief review of nonclassification criteria laboratory assays with potential future diagnostic utility is presented.
Learning Objectives
To understand that the term antiphospholipid is a misnomer: the major autoantigen in APS is β2GPI
To understand that aCL, anti-β2GPI, and LAC triple positivity does not predict thrombotic risk
Clinical features of APS
The core clinical manifestations of the antiphospholipid syndrome (APS) can be divided into thrombotic and obstetrical.1 The former includes both venous and arterial thrombosis and can affect any part of the vascular bed. The most common site of venous thrombosis is in the lower limbs; the most common site of arterial thrombosis is the cerebral circulation.2 The obstetrical manifestations include recurrent first-trimester miscarriages and/or a single second-trimester fetal death and/or early-onset severe preeclampsia.1
Thrombocytopenia, autoimmune hemolytic anemia, heart valve thickening and dysfunction, and livedo reticularis, which are not part of the formal clinical classification criteria for APS, have been associated with patients diagnosed with APS.1,2
In view of the lack of specificity of the clinical manifestations of APS, the laboratory investigations constitute a critical necessity to establish a diagnosis of APS and to allow for classification in clinical APS studies. The laboratory investigations are designed to detect elevated levels of pathologically relevant autoantibodies, which are generically described as antiphospholipid (aPL) autoantibodies (aAbs).1
The accurate laboratory testing for APS carries significant implications for individuals who have suffered from one of the core clinical manifestations. In the setting of venous thrombosis, the implication of being diagnosed with APS is consideration of indefinite anticoagulation, with the associated cumulative risk of bleeding complications. This strategy is distinct to the management plan of non-APS venous thrombosis, although the evidence for this is not strong.3,4 Likewise, in the setting of an individual having an arterial thrombosis such as a stroke, a diagnosis of APS entails consideration for treatment with either an antiplatelet agent such as aspirin or an anticoagulant such as a vitamin K antagonist (VKA; i.e., warfarin).5
The term aPL aAbs is a misnomer. The aAbs that characterize APS do not directly bind to phospholipids. The main APS autoantigen is beta 2-glycoprotein I (β2GPI), an abundant plasma protein that binds to anionic phospholipids.6 The detection of anti-β2GPI aAbs with the anti-β2GPI ELISA has recently been included as part of the laboratory classification criteria for APS (Figure 1).1 There is an extensive body of evidence to suggest that these aAbs may be directly pathogenic.7

Laboratory tests for APS
There are 2 broad categories of assays that are used to diagnose APS: lupus anticoagulant (LAC) assays and ELISAs.
LAC assays
There are several in vitro coagulation-based assays that have as their common point the measurement of the time to clot in vitro of the patient's plasma relative to the time taken by healthy control samples. This is known as a screening test. If prolongation of the patient's clotting time is demonstrated, then 2 further tests are undertaken, first to assess whether the prolonged clotting time is reversed with mixing (1:1 ratio) the patient's plasma with pooled plasma from healthy controls and second to assess whether the prolongation in clotting time seen in the screening test is reversed with the addition of excess anionic phospholipid. The latter test is known as a confirmatory test. These in vitro coagulation-based assays are collectively termed LAC assays. A plasma sample is defined as testing positive for LAC if it has a prolonged in vitro coagulation time that is not reversed with mixing studies, but is reversed when extra phospholipid is added.10 There are several different types of LAC assays available. The dilute Russell viper venom test (dRVVT) assay uses a component of the venom derived from the Russell viper in conjunction with dilute phospholipids to activate factor X to factor Xa. In contrast, the activated partial thromboplastin time (aPTT), the kaolin clotting time (KCT), and the silica clotting time (SCT) assays activate the contact activation and common coagulation pathways.11 The International Society of Haemostasis and Thrombosis (ISTH),12 the British Committee for Standards in Haematology (BCSH),13 and the Clinical and Laboratory Standards Institute (CLSI)14 recognize that a single assay that can detect all individuals who are truly positive for LAC does not currently exist, so in their respective guidelines, they recommend that 2 different assays with distinct performance principles be undertaken to detect LAC.15
A recent survey in Australia found that most hematology laboratories perform the dRVVT and aPTT LAC assays and 1/3 also use the KCT method. The SCT assay is used by <5% of laboratories.16 As part of this quality assurance program, the interlaboratory coefficients of variation were determined16 and compared favorably to the ELISA assays used for diagnosing APS.17 The LAC phenomenon is due to the immunoglobulin-containing fraction in the plasma. The specific aAbs responsible for LAC activity are anti-β2GPI and anti-PT.18,19 Although both anti-β2GPI and anti-PT can be responsible for a positive LAC result, it has been noted that it is LAC due to anti-β2GPI aAbs that most strongly correlates with thrombosis.20 The emphasis placed on the utility of the dRVVT assay as an important LAC screening test in the ISTH, BCSH, and CLSI guidelines derives from studies suggesting that this assay may be more specific in detecting LAC due to anti-β2GPI aAbs compared with the KCT assay.21,22 Consistent with this, some studies have found that a positive dRVVT assay correlates more closely with thrombosis than positivity of the KCT assay.23 It has been noted that the aforementioned studies have been relatively small in scope and that variability in diagnostic performance may exist for reagents from different manufacturers.15
When it comes to considerations as to which LAC screening assay to pair with the dRVVT assay, incorporation of the KCT test is declining from ∼50% of laboratories in Australia in 2009 to 35% in 2011.16 The ISTH (2009) recommendations advise that the aPTT screening assay be performed, with silica as the activator, and low phospholipid content to increase the sensitivity of the assay.12 Ellagic acid as an activator is not advised due to its insensitivity for LAC.12 Kaolin as an activator has been noted to be problematic in automated coagulometers.12 Although because of its extremely low phospholipid content, the KCT assay is very sensitive as a screening test for LAC, an important reason it has started to go out of favor is that it does not have a readily available confirmatory test.15 This imposes a significant limitation on its specificity. SCT has the advantage of being compatible with automated analyzers using photo-optical clot detectors.15
ELISAs
The other assays used to detect so-called aPL aAbs for diagnosing APS are the ELISAs, specifically the anti-β2GPI and the anticardiolipin (aCL) ELISAs.1 These assays can be purchased or can be constructed in-house. They involve coating of an ELISA plate with either β2GPI or the anionic phospholipid cardiolipin, adding the patient's serum at a prespecified dilution (1:50), followed by the application of a secondary labeled antibody that allows quantitation of the bound IgG or IgM isotypes.24 The aCL ELISA detects the presence of antibodies that bind directly to cardiolipin and other anionic phospholipids. It also detects aAbs that bind to β2GPI that has been coated onto the anionic phospholipid surface.25 The aCL ELISA is theoretically less specific than the anti-β2GPI ELISA in diagnosing APS because the former also detects nonspecific antibodies that are present in an individual's plasma as a result of diverse infections.26 One approach to circumventing the problem of distinguishing between APS-relevant aAbs and nonspecific infection-related Abs is the recommendation to retest for aPL Abs at least 12 weeks after the patient initially tests positive.1 APS-related aAbs are more likely to remain persistently positive. Although aAbs detected by the anti-β2GPI ELISA and the LAC assays theoretically are less likely to detect non-APS-related aAbs than the aCL ELISA, it is still recommended that, if initially positive in a patient suspected of having APS, that these assays be repeated after 12 weeks.1 This recommendation acts as a safety guard against over diagnosing APS due to false-positive initial readings.
β2GPI: the main APS autoantigen
β2GPI is a protein composed of 326 amino acids. It has 5 domains, domain I at the N-terminus of the protein, through to domain V at the C-terminus. Domain V also contains a positively charged lysine-rich region, in close proximity to a hydrophobic loop region, which enables β2GPI to bind negatively charged phospholipids.27 The affinity of binding is dependent on the nature and concentration of the anionic phospholipid and the ionic strength of the buffer solution when the β2GPI is coated onto the cardiolipin plate.28 The serine proteases plasmin and factor XIa cleave domain V, which leads to disruption of the hydrophobic loop and the positively charged lysine-rich region, resulting in diminished binding of β2GPI to anionic phospholipids.29-31 A commercial β2GPI preparation has previously been noted to contain the clipped form of β2GPI, with implications for the efficiency of β2GPI binding to the cardiolipin on the ELISA plate.30
Anti-β2GPI autoantibody characteristics
In fluid phase, the β2GPI molecule can potentially exist in either an open, S-type configuration or a closed loop conformation.27,32 Anti-β2GPI aAbs do not consistently form immune complexes with β2GPI in fluid phase for several reasons. Patient aAbs, unlike mAbs from an immunized source, are of low affinity. The β2GPI molecule, when in the closed circular conformation, hides a cryptic epitope on domain I, thus preventing patient aAbs from binding.32 In contrast, when β2GPI is immobilized on a negatively charged phospholipid surface via domain V, the configuration of β2GPI changes to an open form, exposing the cryptic epitope on domain I to which patient anti-β2GPI aAbs are then able to bind (Figure 2).32 Residues 19, 40, and 43 are critical aAb-binding epitopes in domain I.33 Furthermore, patient anti-β2GPI aAbs bind β2GPI when it is coated above a certain antigen density threshold, emphasizing the critical importance of anti-β2GPI aAb-β2GPI interactions being contingent on divalent binding, as opposed to monovalent binding.34 Significant interlaboratory variability with the performance of the anti-β2GPI ELISA may reside in the inconsistent exposure of residues 40 and 43 on domain I when β2GPI is coated onto various commercial ELISA plates.35

Representation of APS autoantibody binding. (A) aCL assay. Shown is the autoantibody (Y) binding negatively charged (−) phospholipid. (B) β2GPI-dependent aCL assay. Shown is the autoantibody binding to domain I of β2GPI. β2GPI domains are denoted by the numbers 1-5. (C) Anti-β2GPI domain I assay.
Representation of APS autoantibody binding. (A) aCL assay. Shown is the autoantibody (Y) binding negatively charged (−) phospholipid. (B) β2GPI-dependent aCL assay. Shown is the autoantibody binding to domain I of β2GPI. β2GPI domains are denoted by the numbers 1-5. (C) Anti-β2GPI domain I assay.
Considerations for when to test for APS
We recommend testing for APS when an individual has suffered an unprovoked thrombotic event. The pretest probability of the diagnosis being APS is likely to be higher if the individual is noted to have one of the noncore clinical criteria, such as thrombocytopenia, autoimmune hemolytic anemia, livedo reticularis, or a heart murmur not due to an alternate diagnosis. A significant increased percentage of LAC or anti-β2GPI ELISA positivity in premenopausal women with stroke and/or myocardial infarction compared with age- and sex-matched controls has been noted.36,37 Smoking and oral contraceptive use are synergistic risk factors in the presence of LAC positivity.36 In a multinational, prospective, 10-year observational study, the mean age of APS patients at study entry was 42 years.2 Testing for obstetric APS is recommended after the third recurrent first trimester miscarriage (<10 weeks gestation), after one fetal death in the second trimester, or after a pregnancy is complicated by severe preeclampsia before the 34th week of gestation, necessitating delivery.1 Other causes of miscarriage and fetal death also need to be excluded.1
Overview comparing LAC assays with the APS ELISA assays
LAC positivity correlates with the clinical features of APS much more strongly than positivity on the ELISAs alone. LAC-positive individuals have higher titers of anti-β2GPI aAbs compared with individuals who are only positive on the anti-β2GPI and/or aCL ELISA alone.38 A significant proportion of APS patients who are LAC positive have anti-β2GPI aAbs that specifically bind to the 19, 40, 43 epitope on domain I.39 There are likely to be other aAbs that are relevant because there are a significant number of LAC-positive APS patients who are negative on the anti-β2GPI and aCL ELISAs.40
Issue of triple positivity
One observational study focusing on women with obstetric APS (with or without a history of previous thrombosis) has suggested that women who test positive on one of the LAC assays and on the anti-β2GPI and aCL ELISAs, so-called “triple-positive” patients, are at higher risk for future thrombotic events and obstetric complications than women with obstetric APS who are not triple positive.38 This study involved 53 women with obstetric APS, of whom 37 were persistently positive on the aCL and anti-β2GPI ELISA and 16 were also positive on one of the LAC assays.38 In a much larger observational study conducted over 10 years, a comparison of thrombotic and obstetric outcomes was undertaken between 513 women with purely obstetric APS (no history of thrombosis at the time of inclusion into the study) and 791 women with a history of 3 recurrent miscarriages before 10 weeks gestation (or one fetal death after 10 weeks gestation) and negative for aPL Abs and 279 women carrying a genetic thrombophilia polymorphism.41,42 Approximately 30% of the APS women were triple positive. The annual rates of deep vein thrombosis (1.46%), pulmonary embolism (0.43%), and cerebrovascular events (0.32%) were significantly higher in aPL aAb-positive women than in the other groups despite the patients in the former group being on low-molecular-weight heparin and low-dose aspirin.41 Being LAC positive was the main predictor for unprovoked proximal or distal DVT and superficial vein thrombosis.41 The thrombotic risks attributable to triple positivity were concordant with the risks conferred by LAC positivity alone, with the exception of pulmonary embolus, for which triple positivity but not LAC alone was a predictor.41 Triple positivity did not predict miscarriage, fetal loss, preeclampsia, premature birth, small for gestational age, or any other placenta-mediated complications.42 In a prospective observational study involving multiple medical centers, the role of aPL aAbs as predictors of adverse pregnancy outcomes were assessed in a cohort of patients that included individuals with established APS (obstetric and/or thrombotic) and/or systemic lupus erythematosus at the time of entry into the study and healthy controls.43 Simultaneous aCL, anti-β2GPI, and LAC did not predict an adverse pregnancy better than did LAC alone.43
Using samples collected during a large, multicenter, population-based, case-control study that enrolled women <50 years of age (the RATIO study), it was noted that the presence of LAC and any additional aPL aAb subpopulation (aCL, anti-β2GPI, and anti-PT ELISA) did not affect the risk of myocardial infarction or ischemic stroke compared with the risk in patients with only LAC.36
A prospective study has assessed the rate of thrombotic events in a cohort of 104 individuals who were initially asymptomatic at study entry who had been found to be triple positive.44 These individuals had been tested for aPL aAbs either because they were noted to have prolonged clotting times during routine coagulation profile testing or they had a history of autoimmunity. Patients with a history of thrombosis or pregnancy complications consistent with APS were excluded from the study.44 The mean age of the study participants was 45 years and 79% were female. There was no control arm in this prospective study. The rate of thrombosis was 5.3% per year. In summary, LAC positivity alone appears to predict thrombotic and obstetric risk as well as triple positivity.
Review of the laboratory guidelines for LAC and ELISA assays
For LAC assays, comprehensive guidelines have been published that discuss preanalytical, analytical, and postanalytical variables.12-14 The similarities and points of difference and rationale behind the differences have also been reviewed in detail (Table 1).15 Likewise, guidelines for laboratories to follow have been published for the performance of the APS ELISAs that are part of the classification criteria (Table 2).24,45
Summary of commonalities and contrasts between recent ISTH, BCSH, and CLSI guidelines for LAC detection
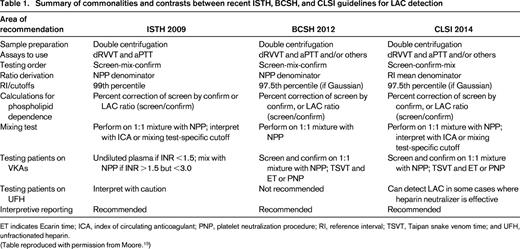
ET indicates Ecarin time; ICA, index of circulating anticoagulant; PNP, platelet neutralization procedure; RI, reference interval; TSVT, Taipan snake venom time; and UFH, unfractionated heparin.
(Table reproduced with permission from Moore.15 )
Summary of recommendations for aCL and anti-β2GPI testing
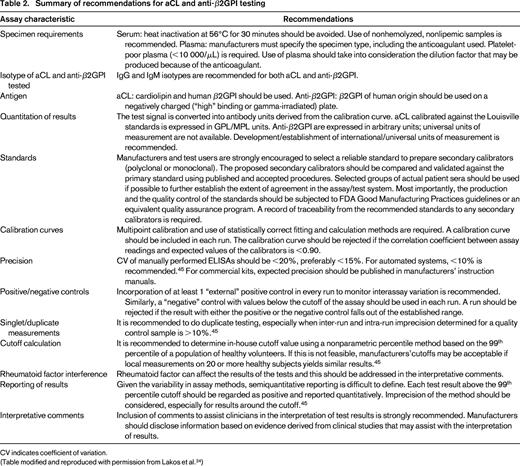
CV indicates coefficient of variation.
(Table modified and reproduced with permission from Lakos et al.24 )
LAC assays
Preanalytic considerations
The presence of platelets in the plasma can affect the interpretation of LAC assays, potentially leading to a false-negative result due to anionic phospholipid expressed on the platelet surface. It is recommended that plasma be rendered platelet poor via a process of double centrifugation.15 Ultracentrifugation (>5000g) may generate microparticles, which can also interfere with the LAC assay; therefore, it is also discouraged. Previously, filtration of the plasma sample through 0.22 μm filters to remove platelets was a procedure undertaken to circumvent the problem of platelet contamination of the plasma sample. This procedure may cause loss of VWF and consequently factor VIII, leading to artificial prolongation of coagulation tests responsive to factor VIII, namely aPTT.11 For this reason, plasma filtration is no longer recommended.
Testing for LAC during an acute illness is discouraged because factor VIII or C-reactive protein levels may be elevated; the former can mask the LAC-screening test, leading to a false-negative result, and the latter may lead to a false-positive screening test result.46
Reference intervals and cutoffs
The ISTH 2009 guidelines recommend that the cutoff for considering a LAC screening assay to be positive be based on determining locally the reference intervals specific to the reagent–analyzer pairings in use and using the 99th percentile.12 This equates to the reference interval mean ± 2.3 SDs for data that has a Gaussian or near-Gaussian distribution. As discussed by Moore,15 this is a controversial decision because it may require a minimum of 120 normal donors, rather than the recommended 40 by ISTH (2009), and an even greater number for nonnormally distributed data. CLSI (2014) recommends deriving the reference interval using the mean ± 2 SDs, with the cutoff for positive being greater than the 97.5th percentile.14 BCSH (2012) provides practical advice regarding validating cutoffs that have been previously established either by the reagent manufacturer or from a different analyzer using just 20-60 healthy donors regardless of whether the 97.5th or 99th percentile is used.13 Concerns about more false-positive LAC being detected if the 97.5th percentile is used rather than the 99th percentile are countered with the insight that a statistical outlier who has a prolonged clotting time on the screening test not related to LAC will also have a similarly prolonged clotting time on the confirmatory test, and thus will not generate a false- positive interpretation. The corollary of this is that using the 99th percentile may lead to more false negatives and a failure to detect clinically meaningful LAC-positive patients.15
Mixing tests
The sequence of tests performed by laboratories to determine LAC positivity is another topic of contention between different guidelines.15 Traditionally, the sequence has been to perform the screening test, then the mixing studies, and, if the mixing studies demonstrate lack of correction, to then proceed to a confirmatory test for LAC. Concerns have been raised at having the mixing studies performed at the second stage and proceeding only if they demonstrate a lack of correction. The reasoning is that mixing studies may dilute out the in vitro effect of clinically relevant aPL aAbs and thus may systematically bias results toward false-negative readings for LAC if a decision to proceed to the confirmatory test is stopped at this point. It has been reasoned that, in the context that other types of coagulation disturbances have been excluded by undertaking routine coagulation screening, including PT time, thrombin time, and aPTT using a LAC-unresponsive aPTT reagent, that testing positive at the screening and confirmatory test stages, even if the mixing studies are negative, is adequate to consider the sample LAC positive.15 CLSI (2014), in contrast to ISTH (2009), has recommended the reordering of the sequence of testing for LAC to screening, confirmatory testing, and then mixing studies.12,14 In this schema, the utility of performing the mixing studies is in cases where there is prolongation of time to clotting in the screening test without a corresponding correction in the confirmatory test in the absence of a readily identifiable alternate coagulation disturbance when routine coagulation studies are undertaken. This situation may arise either when there is an undiagnosed coagulopathy or if the aPL aAbs are sufficiently potent to fully prevent the reagent and excess phospholipid in the confirmatory test from correcting the prolongation in clotting time. Mixing the patient's plasma at a 1:1 ratio with normal pooled plasma (NPP) and then undertaking the screen and confirmatory tests may allow the detection of LAC by diluting the potency of the aPL aAbs.
The index of circulating anticoagulant is one option for assessing mixing study results; the other is to determine a mixing-study-specific reference interval that will allow the establishment of cutoff values.15
Ratios
The conversion of clotting times for screen and confirm tests to normalized ratios using the value of NPP is advocated by the ISTH (2009) and BCSH (2012).12,13 This practice has been noted to improve the performance of the LAC assays, reducing inter-assay and intra-assay variation by minimizing differences in operator and/or analyzer performance, reagent quality, and stability issues and NPP clotting time variation. As a point of contrast, CLSI (2014) advises to normalize against the reference interval mean clotting time rather than the NPP value.14 It is argued that not all NPP batches generate the same clotting times, with different reagents for the same test type, which can systematically bias readings toward false-positive or false-negative results if an NPP value is at the extreme of the reference interval.15 It is recommended that the reference interval and its mean be determined over several days by analyzing normal donors, which will account for innate technique variability. NPP samples can be used as normal controls to identify sudden analytical difficulties.15
Confirmatory tests
All 3 guidelines are in agreement that the confirmatory test should be based on the same screening test that was positive to avoid problems with loss of specificity and false-negative interpretations by doing otherwise. The concept is to use the framework of the screening test but, in addition, to add a higher concentration of phospholipid, which can take the form of either bilayer or hexagonal (II) phase phospholipid. The use of freeze/thawed platelets as a source of phospholipid is not encouraged due to inconsistency in the amount of phospholipid between batches.
Reporting of the confirmatory test can be with the use of the following formula: percentage correction of ratio = [(screen ratio − confirm ratio)/(screen ratio)] × 100.
The BCSH (2012) advocates that a correction of 10% or more be interpreted as consistent with LAC positivity.13 ISTH (2009) advises that the cutoff should be locally derived.12 Conversely, CLSI (2014) recommends assessing deltas when the hexagonal phase phospholipid neutralization test is used in the context of the aPTT confirmatory assay.14 When either the dRVVT or the SCT assays are used with paired screen and confirm reagents, respectively, the normalized screen to confirmatory ratio is recommended:15 normalized screen:confirm ratio = screen ratio/confirm ratio.
Testing patients on anticoagulants
LAC testing in the setting of an individual taking a VKA is not an absolute contraindication, although it is universally acknowledged by all 3 guidelines that it is not optimal to test for LAC while the patient is taking VKA.12-14 BCSH (2012) advocates brief discontinuation of VKA therapy in the context that testing for LAC will determine whether the patient be considered for long-term anticoagulation.13 If LAC testing must be done while the patient is on VKA therapy, then the recommendations of the guidelines do differ slightly in some respects. If the international normalized ratio (INR) is <1.5, ISTH (2009) suggests that the performance of dRVVT and aPTT assays be undertaken on undiluted plasma. If, however, the INR is between 1.5 and ≤3, then the tests should be undertaken using 1:1 mixtures with NPP.12 BCSH (2012) and CLSI (2014) also advocate the use of 1:1 mixture with NPP when performing the screening and confirmation tests, but do not restrict this practice to individuals with an INR ≤3 alone.13,14 All guidelines acknowledge the possibility of a false-negative result due to the dilution effect of the mixing studies. Testing on undiluted plasma in the context of an INR ≥1.5 is susceptible to false positives and false negatives. Taipan snake venom time can be a useful screening test for LAC in patients taking VKA, and Ecarin time can serve as a confirmatory test in this context.15
Unlike VKA, which leads to coagulation factor deficiencies that can be corrected with mixing studies, thus allowing detection of LAC positivity, the coagulation effects caused by direct thrombin inhibitors (dabigatran and argatroban) and the fXa inhibitor (rivaroxaban) cannot be reversed by mixing studies. Therefore, these medications interfere with all LAC assays and thus introduce a significant risk of false-positive results.15 Taipan snake venom time (screening) and Ecarin time (confirmation) can be useful in detecting LAC in individuals on rivaroxaban because both venoms are PT activators and thus are unaffected by fXa inhibition.15
Testing for LAC in the presence of heparin is discouraged, although the problems can be circumvented to some degree with the use of heparin neutralizers, which can be added to the sample plasma or can be found in most commercial dRVVT reagents.15 It is important that an assessment be undertaken to ensure that all of the heparin has been neutralized before proceeding to LAC testing.15
Reporting
It is encouraged by the various guidelines that a definitive summary statement be given when reporting the results of the LAC assay results whether LAC is present or not detected. Terms such as borderline or weak positive are discouraged.15
Solid-phase assays
Preanalytical variables
Most ELISA systems recommend the use of serum samples.24 If plasma is used, it is important to ensure that it is platelet poor and a correction is made for the volume of anticoagulant in the collecting tube.24
There are several variables that interfere with the optimal performance of the ELISA assays, including hemoglobin, bilirubin, and lipemia.47 IgM rheumatoid factor is another variable that has been noted on occasion (although not consistently) to cause interference in IgM aCL and IgM anti-β2GPI ELISA assays and can lead to false-positive readings.47
Calibrators and standard curves
Interlaboratory variability seen with the performance of the aCL and anti-β2GPI ELISAs is due to the lack of uniformity in reference material for calibration.17,46 The Louisville standards were the original calibrators that were intended to introduce an international standard for the aCL assays.24 However, the issue is that they are a finite resource of well characterized, patient-derived, polyclonal calibrators, with the inherent limitations to widespread availability that this entails.46 Three generations of polyclonal calibrators have subsequently been derived from this original set of standards and have been widely distributed.46 It has been noted that these reference calibrators are not used for routine day-to-day purposes, but rather are used to assign calibrant units to calibrators developed by manufacturers for their own kits.46 The results of aCL ELISA results are expressed in IgG phospholipid (GPL) and IgM (MPL) units. One GPL or MPL unit is defined as the cardiolipin-binding activity of 1 μg/mL of affinity-purified polyclonal IgG or IgM aCL antibody.24
One way of overcoming limitations in uniform calibrator availability was the development of the Sapporo standards, IgM and IgG isotopes that are chimeric mAbs against β2GPI, designated EY2C9 and HCAL, respectively.46 They are distributed by the Centers for Disease Control and Prevention through the Autoantibody Standardization Committee (http://asc.dental.ufl.edu). The advantage of unlimited supply and better reproducibility over the long term is countered by disadvantages such as not behaving as patient-derived antibodies (mAbs are high affinity, patient aAbs are of low affinity) and, furthermore, mAbs do not capture the diverse specificities of aPL aAbs present in APS.46 Assays that use these mAbs are reported in protein concentration units (micrograms per milliliter); however, these units have not been cross-validated against GPL/MPL units, thus complicating the issue of quantitation.24 In addition, universal units of measurement are not available for anti-β2GPI assays and a variety of other arbitrary units are used, including units per milliliter, standard IgG and IgM units, nanograms per milliliter, optical density values etc. Strong recommendations have been made for a concerted effort to establish international units for measurement of anti-β2GPI aAbs detected by anti-β2GPI ELISA assays to facilitate uniformity and comparability of results among different assays.24,45
Cutoffs
In the International Consensus Guidelines on Anticardiolipin and Anti-β2GPI testing, it is recommended that the reference range be established by the nonparametric percentile method in view of the fact that autoantibody values do not tend to follow a Gaussian distribution.24,45 A minimum of 120 reference subjects, taking into consideration such variables as age and type of population most representative for each laboratory, are used to establish the reference interval.45 Alternatively, for small laboratories that do not have the resources to establish their own reference range, it is deemed appropriate to undertake a verification process to affirm the manufacturers' reference ranges using a small number (minimum 20) of appropriately selected reference individuals using professionally and statistically sound protocols, as outlined in the CLSI C28-A3 document.48
For the aCL ELISA, levels of IgG and/or IgM greater than the 99th percentile or >40 GPL or MPL (in cases where the Louisville calibrators or their derivatives are used) are included as classification criteria for APS.1 For the anti-β2GPI ELISAs, the 99th percentile is used as the cutoff. The use of the 99th percentile seems to be more sensitive for APS classification purposes than the >40 GPL cutoff.46 IgM aAbs are less often associated with thrombosis than IgG.46 Given the variability in assay methods, semiquantitative reporting is difficult to define. Each test above cutoff should be regarded as positive and reported quantitatively.45
Nonclassification criteria laboratory assays
There is a significant subset of LAC-positive APS patients who do not have anti-β2GPI aAbs.40 The nature of the aAbs responsible for this subset of LAC positivity in APS is a matter of ongoing investigation. Several researchers have suggested that these aAbs may be targeting PT complexed to phosphatidylserine (PS).49 The older generation of anti-PT ELISAs were not designed to specifically detect aAbs that target the PS/PT complex, in which it is thought that the PT molecule undergoes conformational change, perhaps exposing a cryptic epitope. There has been a flurry of activity in the development of ELISAs that detect aAbs targeting the PS/PT complex, with some promising results with regard to their potential diagnostic value.50 Larger studies involving multiple centers and, preferably, multinational research collaborations will be necessary before accepting the clinical utility of such findings. Several other active areas of ongoing research based on promising emerging data include determining the diagnostic utility of a novel domain I–specific anti-β2GPI ELISA and the relevance of IgA anti-β2GPI aAbs.51
Disclosures
Conflict-of-interest disclosures: The authors declare no competing financial interests. Off-label drug use: None disclosed.
Correspondence
Steven A. Krilis or Bill Giannakopoulos, Department of Infectious Diseases, Immunology and Sexual Health, St. George Hospital, University of New South Wales, 2 South Street, Kogarah, Sydney NSW 2217, Australia; Phone: 61-2-9113-2955; Fax: 61-2-9113-3980; e-mail: s.krilis@unsw.edu.au or bill.giannakpoulos@unsw.edu.au.
