

T-cell receptor (TCR) therapies are a promising modality for the treatment of cancers, with significant efforts being directed toward acute myeloid leukemia (AML), a particularly challenging disease. Chimeric antigen receptor (CAR) T cells targeting single surface antigens have shown remarkable efficacy for B-cell lymphoblastic leukemia, lymphomas, and multiple myeloma. However, AML presents formidable obstacles to the effectiveness of CAR T cells because of the widespread expression of heterogenous leukemia immunophenotypes and surface antigen targets additionally present on normal myeloid cells. TCR therapies are an evolving field of cell therapies that allow targeting intracellular antigenic peptides presented via HLA molecules. The development of TCR therapy for AML is progressing rapidly through preclinical research and successful clinical trials. This review specifically explores the antigens targeted in AML, the diverse methodologies and strategies used in TCR identification, and preclinical TCR T-cell development. The review also discusses innovative molecular designs to improve functional efficacy, mitigate safety concerns, and overcome HLA restrictions. Specific outcomes of early clinical trials targeting important antigens Wilms tumor gene 1, preferentially expressed antigen in melanoma, and minor histocompatibility antigen HA-1 are also highlighted. Ultimately, this review underscores why TCR therapy is poised to become an indispensable component of AML immunotherapy.
Adoptive T-cell therapies for AML
Acute myeloid leukemia (AML) is a hematological malignancy arising from aberrant transformation of myeloid lineage cells. It can affect both young children, constituting 18% of pediatric leukemias, as well as adults, with a median age at diagnosis of 68 years.1,2 Younger, fit patients (aged ≤70 years) are typically treated with intensive chemotherapy, and if deemed at high risk of relapse, would undergo allogeneic hematopoietic stem cell (allo-HSCT) transplant while in remission. The long-term survival rates for pediatric cohorts are ∼70%, reducing to just over 50% in patients aged between 18 and 60 years.3,4 Intensive treatments such as chemotherapy and allo-HSCT are not suitable for most older patients because of physiological frailty and inherent differences in genetic drivers of AML. Although hypomethylating agents and Bcl-2 inhibitors have improved outcomes in this group, the 5-year overall survival remains dismal at 27%.5 Overall, 10% to 45% of patients achieve sustained remission, with older patients performing worse than younger patients.6 In recent years, novel immunotherapeutic approaches such as adoptive T-cell therapies are being investigated for AML. This has led to promising advances in developing new treatment options, although key challenges remain, and no AML-directed cellular therapies aside from allo-HSCT are yet routinely available.
After the discovery of the role of immune cells in tumor suppression,7 the earliest form of adoptive cell therapy for malignant diseases were tumor-infiltrating lymphocytes or circulating cytotoxic lymphocytes (CTLs). Tumor-infiltrating lymphocytes and CTLs were isolated from patients or donors, expanded ex vivo, sometimes with additional antigenic stimulus to expand antitumor T-cell clones, and reinfused back into the patient.8,9 For hematological malignancies such as AML, there have been a handful of trials using HLA-matched ex vivo expanded CTLs, with favorable clinical responses observed.10-12 Multiantigen-targeting CTLs to improve graft-versus-leukemia have shown minimal toxicity and favorable response13 and can even persist 1 year after infusion.14 However, generation of a sufficient polyclonal CTL product is not always possible. Naturally occurring AML tumor antigen–specific T cells (such as against Wilms tumor gene 1 [WT1] or preferentially expressed antigen in melanoma [PRAME]) in healthy donors are infrequent (<0.05% in circulation) and corresponding T-cell receptors vary in binding affinity, leading to variability in response. Furthermore, extended ex vivo stimulation could lead to exhaustion and further hinder efficacy in vivo, hindering their widespread use.
The development of chimeric antigen receptor (CAR) T cells presented a unique solution to these challenges, by modifying peripheral blood T cells to express a synthetic chimeric receptor (CAR), allowing for generation of a more uniform, reliable product with streamlined manufacture processes.15-18 The CAR molecule recognizes surface antigens in a HLA-independent manner, which results in T-cell activation, cytokine release, and targeted cytotoxicity. CAR T-cell therapy has had great clinical success in large B-cell lymphomas, mantle-cell lymphoma, B-cell acute lymphoblastic leukemia, and multiple myeloma. There are currently 6 US Food and Drug Administration–approved products targeting either CD19 or B-cell maturation antigen.19 Unfortunately, despite extensive preclinical work,20 these successes have yet to be translated to AML, because of, in part, lack of suitable surface antigens. Some promising targets (eg, CD33 and CD123) are shared with healthy hemopoietic cells, and require rescue allo-HSCT because of myelosuppression caused by treatment, increasing potential treatment toxicities.21 Moreover, there is heterogeneity in antigen expression between patients and also within the malignant T-cell population of each individual patient.22 One of the challenges for AML therapies remains in identifying the ideal targetable antigens.
TCR therapy is an alternative adoptive cell therapy that involves the introduction of TCR sequences into T cells to redirect specificity toward a target antigen. The TCR is a macromolecular multisubunit structure comprising of antigen-recognizing α and β subunits that combine with signaling subunits of the CD3 molecule (Figure 1). Binding of the antigen–HLA complex allows the recruitment of kinases, phosphorylation of key domains that result in downstream signaling, activation, and functional response.23 TCRs differ from CARs in target recognition (requiring HLA-dependency), sensitivity, and signaling (Table 1). The nature and context of the antigen informs the choice of using either a CAR or TCR for targeting. For example, in contrast to CARs that target surface antigens, TCRs can recognize a larger pool of intracellular antigens through antigenic peptides that have been processed internally and presented on the HLA complex on the cell surface. Native TCRs undergo thymic selection, and are highly discriminatory with 100-fold higher antigen sensitivity than CARs.33 Most TCRs are highly specific, recognizing antigenic peptides that are generally presented on only a single HLA subtype; HLA molecules are polymorphic, with over 1000 alleles reported. HLA-A∗02:01 and HLA-A∗24:02 alleles are present at higher frequencies across the global population.35 Although TCR therapy can be limiting because of its inherent HLA restriction, the potential to overcome constraints of CTL therapy is attractive, especially in the treatment of AML, which has numerous intracellular proteins with immunogenic peptide epitopes presented on various HLAs. Thus, this review will highlight recent developments in TCR therapy development for AML.

TCR and CAR. TCR αβ subunits combine with γ, δ, ε, and ζ subunits of the CD3 to form the multisubunit TCR complex. Peptide-HLA binding to the TCR αβ combined with the other subunits and with either CD4 or CD8 coreceptor initiates downstream signaling, T-cell activation, and effector functioning. Proteins involved in signaling such as LCK and ZAP70 are indicated and the activating motifs within the subunits are represented as horizontal bars. A typical CAR molecule comprises an antigen-specific scFv from a monoclonal antibody linked to a spacer, costimulatory domain (CD28 or 4-1BB shown in this example) and signaling CD3ζ domain. Figure created with BioRender.com: Gore S. (2025); https://BioRender.com/o91f055
TCR and CAR. TCR αβ subunits combine with γ, δ, ε, and ζ subunits of the CD3 to form the multisubunit TCR complex. Peptide-HLA binding to the TCR αβ combined with the other subunits and with either CD4 or CD8 coreceptor initiates downstream signaling, T-cell activation, and effector functioning. Proteins involved in signaling such as LCK and ZAP70 are indicated and the activating motifs within the subunits are represented as horizontal bars. A typical CAR molecule comprises an antigen-specific scFv from a monoclonal antibody linked to a spacer, costimulatory domain (CD28 or 4-1BB shown in this example) and signaling CD3ζ domain. Figure created with BioRender.com: Gore S. (2025); https://BioRender.com/o91f055
Differences in TCRs and CARs
| TCR | CAR | |
|---|---|---|
| Target | Intracellular antigen (recognition of peptide/HLA complex), HLA restricted | Surface antigen (conventionally, not always) |
| Intracellular signaling domains | Formation of CD3ελ, CD3εδ heterodimers, and CD3ζ homodimer, 10 ITAMs | Conventionally some combination of linked CD28 or 4-1BB and CD3ζ, 3 ITAMs |
| Coreceptor requirement | Yes (CD4, CD8) | Not required |
| Activation threshold | 1-3 molecules on target cell24,25 | >100 molecules on target cell26 |
| Sensitivity | Higher antigen sensitivity27-29 | Lower antigen sensitivity,27-29 up to 100-fold less at low-antigen levels30,31 |
| Affinity | Generally lower (allowing for serial triggering),32 in the mM range | Slightly higher,33 in the nM range |
| Immune synapse formation | Highly organized, 5-10 min formation time34 | Disorganized, <2 min formation time34 |
| Downstream activation/phosphorylation | Stronger activation28 | Weaker activation28 |
| TCR | CAR | |
|---|---|---|
| Target | Intracellular antigen (recognition of peptide/HLA complex), HLA restricted | Surface antigen (conventionally, not always) |
| Intracellular signaling domains | Formation of CD3ελ, CD3εδ heterodimers, and CD3ζ homodimer, 10 ITAMs | Conventionally some combination of linked CD28 or 4-1BB and CD3ζ, 3 ITAMs |
| Coreceptor requirement | Yes (CD4, CD8) | Not required |
| Activation threshold | 1-3 molecules on target cell24,25 | >100 molecules on target cell26 |
| Sensitivity | Higher antigen sensitivity27-29 | Lower antigen sensitivity,27-29 up to 100-fold less at low-antigen levels30,31 |
| Affinity | Generally lower (allowing for serial triggering),32 in the mM range | Slightly higher,33 in the nM range |
| Immune synapse formation | Highly organized, 5-10 min formation time34 | Disorganized, <2 min formation time34 |
| Downstream activation/phosphorylation | Stronger activation28 | Weaker activation28 |
ITAMs, immunoreceptor tyrosine-based activation motifs.
Preclinical development of TCR T-cell therapy in AML
Antigen selection
A range of AML associated antigens have been targeted with TCR T-cell therapy. Figure 2 provides an overview of the preclinical development methodologies. The studies outlined in Table 2 are limited to antigens, which, to date, have been targeted with TCR T cells in AML. Suitable antigens possess desirable characteristics, including immunogenicity and cancer-specific or cancer-associated expression.57

Pipeline of TCR T-cell preclinical development. (A) Antigens can either be overexpressed self-antigens seen on normal tissues or those that belong to the cancer testis antigen family with restricted expression; neo-antigens arising from mutations and gene rearrangements; or those belonging to the minor histocompatibility complex. (B) Donor PBMCs containing either auto- or allo-HLA–restricted TCR-expressing T cells can be stimulated with antigenic peptides; and activated or reactive T cells can be identified, isolated, and expanded in the laboratory. (C) Sequencing of reactive cells identifies the precise αβ sequences of the TCR that are introduced into T cells after modifications to ensure proper pairing. Newly generated TCR T cells are tested for function and efficacy against tumor cells and tested for specificity and safety using in silico and experimental analyses. CDX, cell line-derived xenograft; DCs, dendritic cells; PDX, patient-derived xenograft. Figure created with BioRender.com: Gore S (2025); https://BioRender.com/o91f055
Pipeline of TCR T-cell preclinical development. (A) Antigens can either be overexpressed self-antigens seen on normal tissues or those that belong to the cancer testis antigen family with restricted expression; neo-antigens arising from mutations and gene rearrangements; or those belonging to the minor histocompatibility complex. (B) Donor PBMCs containing either auto- or allo-HLA–restricted TCR-expressing T cells can be stimulated with antigenic peptides; and activated or reactive T cells can be identified, isolated, and expanded in the laboratory. (C) Sequencing of reactive cells identifies the precise αβ sequences of the TCR that are introduced into T cells after modifications to ensure proper pairing. Newly generated TCR T cells are tested for function and efficacy against tumor cells and tested for specificity and safety using in silico and experimental analyses. CDX, cell line-derived xenograft; DCs, dendritic cells; PDX, patient-derived xenograft. Figure created with BioRender.com: Gore S (2025); https://BioRender.com/o91f055
Preclinical studies testing TCR T cells for AML
| Reference | Target epitope | HLA | Donor pool | Pool type | Specific stimulation | Isolation | Sequencing | TCR modification | Additional elements | Transduction | Recipient cells | Testing methods | Testing against |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Overexpressed antigens | |||||||||||||
| Ruggerio et al36 | WT137-45 (VLDFAPPGA) | A∗02:01 | Healthy | Not specified | Direct peptide loading | CD137 | RACE PCR | - | CRISPR/Cas9 knockout of endogenous TCR | LV/AAV | Healthy donors Patient T cell | Flow cytometry cytokine release and cytotoxicity assays Live-cell imaging Luciferase assay | Cell lines, modified HLA expression (K562, 697, OCI-AML3) EBV-LCLs Peptide-loaded T2 Primary AML Healthy cells PDX NOD mouse model |
| Van Amerongen et al37 | WT1 (VLDFAPPGA) (ALLPAVPSL) (VLDFAPPGASAY) (TPYSSDNLY) | A∗02:01 A∗02:01 A∗01:01 B∗35:01 | Healthy | Allo | - | Multimer single-cell clone | PCR | Murine constant Cysteine modified | - | RV | Healthy donors | ELISA Chromium 51 | Cell lines, modified HLA/antigen expression, peptide loaded (Raji) EBV-LCLs Healthy cells |
| Lahman et al38 | WT137-45 (VLDFAPPGA) | A∗02:01 | Healthy | Not specified | Peptide-loaded DCs | Tetramer | RACE PCR | - | CD8 coreceptor | LV | Healthy HLA-A∗02:01 donors | Flow cytometry intracellular cytokine release assay | Primary AML Cell lines, modified HLA/antigen expression (K562) PHA blasts, peptide loaded EBV-LCLs CDX NOD and NSG mouse model |
| Xue et al39 | WT1126-134 (RMFPNAPYL) | A∗02:01 | Healthy | Allo | Peptide-loaded T2 cells | Vβ2+ and CD8+ (magnetic bead and FACS) | PCR | - | - | RV | HLA-A2+ Healthy Donor T cells Patient T cells | Chromium 51 IFNγ release | T2 peptide–loaded cells Cell lines (697, BV173, Lama81, Kyo-1) CDX NOD/SCID mouse model |
| Arber et al40 | Survivin96-10497M (LMLGEFLKL) | A∗02:01 | Healthy | Auto | Peptide-loaded DCs | Single-cell clone testing | 5′ RACE PCR | Murine constant | - | RV | Healthy Donor CD8+ cells | Chromium 51 ELISPOT CFU assay | Cell lines (BV173, U266, K562, HL-60) Primary AML CDX NSG mouse model |
| Sandri et al41,42 | TERT865–873 | A∗02:01 | Patients (B-CLL) Mouse CTLs | Auto | Peptide-loaded DCs | Single-cell clone testing | PCR | - | - | RV | Healthy donor PBMCs | ELISA flow cytometry cytotoxicity activity assay | Cell lines (THP1) Peptide-loaded PBMCs CDX NOG mouse model |
| Depreter et al43 | TARP(P5L)4–13 | A∗02:01 | Healthy | Auto | Peptide-loaded DCs | Single-cell clone testing | 5′ RACE PCR | Murine constant | - | LV/RV | Healthy donor CD8+ cells | Flow cytometry cytokine release and cytotoxicity assays Chromium 51 Luciferase assay | Cell lines, modified HLA/antigen expression (THP1, LNCaP, KG1a, Molm13, OCI-AML3, HL60) Primary samples |
| Cancer testes antigens | |||||||||||||
| Amir et al44 | PRAME425-433 (SLLQHLIGL) | A∗02:01 | Patient donors (after HSCT) | Auto- and allo-HSCT | - | Tetramer single-cell sorted | RT-PCR | Cysteine modified | - | RV | EBV-specific T cells | ELISA Chromium 51 | Peptide-loaded T2 cell lines, modified HLA/antigen expression (K562, COS) EBV-LCLs Primary samples Healthy cell subsets |
| Kang et al45 Jager et al46 | NY-ESO-1157-165 | A∗02:01 | Healthy | Auto | Primary tumor cell line | Not specified | Not specified | Cysteine-modified CDR3a dual aa substitution | - | LV | Healthy donor PBMCs | ELISA, ELISPOT LDH cytotoxicity assay | Cell lines (U937, HL60, Kasumi-1) CDX NCG mouse model |
| Nagai et al47 | AURKA207-215 (YLILEYAPL) | A∗02:01 | Healthy | Auto | Peptide-loaded DCs | Not specified | 5′ RACE PCR | - | - | RV | Healthy donor T cells | ELISA, ELISPOT Chromium 51 CSFE proliferation assay Luciferase reporter TCR signaling assay | Cell lines, modified HLA expression (GANMO-1, MEG01, KAZZ, OUN-1) Primary AML samples CDX NOG mouse model |
| Spranger et al48 | HMMR (MSFPKAPL) | A∗02:01 | Healthy | Allo | RNA-loaded DCs | CD137 single-cell clones | PCR | Murine constant | - | RV | Healthy donor T cells | Chromium 51 CFU assay | Cell line, modified HLA expression (K562, THP1) Primary AML CDX NSG mouse model HSCs |
| MiHA | |||||||||||||
| Van Loenen et al49 | HA-1 (VLHDDLLEA) | A∗02:01 | Healthy | Not specified | Not specified | Not specified | Not specified | Cysteine modifications | - | RV | Allo-restricted healthy donor T cells | ELISA Cytotoxicity assays | EBV-LCLs Primary AML |
| Dossa et al50 | HA-1 (VLHDDLLEA) | A∗02:01 | Healthy | Auto | Peptide-loaded DCs | Single-cell clones | RACE PCR | Cysteine modifications | CD8 coreceptor iCasp9 safety switch | LV | Healthy donor T cells | Chromium 51 Intracellular cytokine release and CD107 degranulation flow cytometry | T2 peptide–loaded cells Primary AML |
| Pilunov et al51 | HA-1 | A∗02:01 | Healthy | Auto | Peptide-loaded DCs | CD137/multimter | RACE PCR | Murine constant Cysteine modifications | CRISPR-Cas9 knock down of endogenous TCR | LV | Healthy donor CD8+ cells | Fluorescent reporter J76 cell line assay ELISA Flow cytometry cytotoxicity assay | Primary AML cell lines, modified HLA expressed, peptide loaded (K562) Healthy and patient PBMCs |
| Neoantigens | |||||||||||||
| Van der Lee et al52 | mutNPM1 (CLAVEEVSL) | A∗02:01 | Healthy (unsuccessful with AML patients) | Auto | - | Tetramer single-cell clones | PCR | Murine constant Cysteine modification | - | RV | Healthy PBMCs, later CD4/8 purified | ELISA Chromium 51 | Peptide-loaded T2 cells Primary AML CDX NSG mouse model |
| Van der Lee et al53 | mutNPM1 (AVEEVSLRK) | A∗11:01 | Healthy | Auto | - | Tetramer single-cell clones | PCR | Murine constant | - | RV | Healthy PBMCs, later CD4/8 purified | ELISA Chromium 51 | Peptide-loaded T2 cells EBV-LCLs Cell lines, modified HLA expression (K562, OCI-AML2, OCI-AML3) Primary AML CDX NSG mouse model |
| Biernacki et al54 | CBFB-MYH11 fusion (REEMEVHEL) | B∗40:01 | Healthy | Auto | Peptide-loaded DCs | Single-cell clones | RACE PCR | Cysteine modifications | - | LV | Healthy donor T cells | Chromium 51 Flow cytometry–based cytotoxicity and CD107 degranulation assays | Peptide-loaded LCLs Cell lines, modified HLA/antigen expression (NB4, ME-1) PDX MISTRG mouse model |
| Biernacki et al55 | U2AF1Q157R (DFREACCRR) | A∗33:01/03 | Healthy | Auto | Peptide-loaded DCs | Single-cell clones | RACE PCR | Murine constant Cysteine modifications | CRISPR-Cas9 knock down of endogenous TCR | LV | Healthy donor T cells | Chromium 51 Flow cytometry–based cytotoxicity and CD107 degranulation assays | Peptide-loaded LCLs Cell lines, modified antigen expression (TF-1) Primary AML Healthy PBSCs/MCs CDX NSG mouse model |
| Giannakopoulou et al56 | mutFLT3 - D835Y (YIMSDSNYV) | A2 | Healthy | Auto | RNA-loaded DCs | Multimer single-cell clones | RT-PCR | Murine constant Cysteine modifications | - | RV | Healthy donor PBMCs | ELISA Flow cytometry–based activity and cytotoxicity assays | Cell lines, modified HLA expression, peptide-loaded (K562) EBV-LCLs Primary AML Healthy A2+ cells CDX and PDX NSG mouse model |
| Reference | Target epitope | HLA | Donor pool | Pool type | Specific stimulation | Isolation | Sequencing | TCR modification | Additional elements | Transduction | Recipient cells | Testing methods | Testing against |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Overexpressed antigens | |||||||||||||
| Ruggerio et al36 | WT137-45 (VLDFAPPGA) | A∗02:01 | Healthy | Not specified | Direct peptide loading | CD137 | RACE PCR | - | CRISPR/Cas9 knockout of endogenous TCR | LV/AAV | Healthy donors Patient T cell | Flow cytometry cytokine release and cytotoxicity assays Live-cell imaging Luciferase assay | Cell lines, modified HLA expression (K562, 697, OCI-AML3) EBV-LCLs Peptide-loaded T2 Primary AML Healthy cells PDX NOD mouse model |
| Van Amerongen et al37 | WT1 (VLDFAPPGA) (ALLPAVPSL) (VLDFAPPGASAY) (TPYSSDNLY) | A∗02:01 A∗02:01 A∗01:01 B∗35:01 | Healthy | Allo | - | Multimer single-cell clone | PCR | Murine constant Cysteine modified | - | RV | Healthy donors | ELISA Chromium 51 | Cell lines, modified HLA/antigen expression, peptide loaded (Raji) EBV-LCLs Healthy cells |
| Lahman et al38 | WT137-45 (VLDFAPPGA) | A∗02:01 | Healthy | Not specified | Peptide-loaded DCs | Tetramer | RACE PCR | - | CD8 coreceptor | LV | Healthy HLA-A∗02:01 donors | Flow cytometry intracellular cytokine release assay | Primary AML Cell lines, modified HLA/antigen expression (K562) PHA blasts, peptide loaded EBV-LCLs CDX NOD and NSG mouse model |
| Xue et al39 | WT1126-134 (RMFPNAPYL) | A∗02:01 | Healthy | Allo | Peptide-loaded T2 cells | Vβ2+ and CD8+ (magnetic bead and FACS) | PCR | - | - | RV | HLA-A2+ Healthy Donor T cells Patient T cells | Chromium 51 IFNγ release | T2 peptide–loaded cells Cell lines (697, BV173, Lama81, Kyo-1) CDX NOD/SCID mouse model |
| Arber et al40 | Survivin96-10497M (LMLGEFLKL) | A∗02:01 | Healthy | Auto | Peptide-loaded DCs | Single-cell clone testing | 5′ RACE PCR | Murine constant | - | RV | Healthy Donor CD8+ cells | Chromium 51 ELISPOT CFU assay | Cell lines (BV173, U266, K562, HL-60) Primary AML CDX NSG mouse model |
| Sandri et al41,42 | TERT865–873 | A∗02:01 | Patients (B-CLL) Mouse CTLs | Auto | Peptide-loaded DCs | Single-cell clone testing | PCR | - | - | RV | Healthy donor PBMCs | ELISA flow cytometry cytotoxicity activity assay | Cell lines (THP1) Peptide-loaded PBMCs CDX NOG mouse model |
| Depreter et al43 | TARP(P5L)4–13 | A∗02:01 | Healthy | Auto | Peptide-loaded DCs | Single-cell clone testing | 5′ RACE PCR | Murine constant | - | LV/RV | Healthy donor CD8+ cells | Flow cytometry cytokine release and cytotoxicity assays Chromium 51 Luciferase assay | Cell lines, modified HLA/antigen expression (THP1, LNCaP, KG1a, Molm13, OCI-AML3, HL60) Primary samples |
| Cancer testes antigens | |||||||||||||
| Amir et al44 | PRAME425-433 (SLLQHLIGL) | A∗02:01 | Patient donors (after HSCT) | Auto- and allo-HSCT | - | Tetramer single-cell sorted | RT-PCR | Cysteine modified | - | RV | EBV-specific T cells | ELISA Chromium 51 | Peptide-loaded T2 cell lines, modified HLA/antigen expression (K562, COS) EBV-LCLs Primary samples Healthy cell subsets |
| Kang et al45 Jager et al46 | NY-ESO-1157-165 | A∗02:01 | Healthy | Auto | Primary tumor cell line | Not specified | Not specified | Cysteine-modified CDR3a dual aa substitution | - | LV | Healthy donor PBMCs | ELISA, ELISPOT LDH cytotoxicity assay | Cell lines (U937, HL60, Kasumi-1) CDX NCG mouse model |
| Nagai et al47 | AURKA207-215 (YLILEYAPL) | A∗02:01 | Healthy | Auto | Peptide-loaded DCs | Not specified | 5′ RACE PCR | - | - | RV | Healthy donor T cells | ELISA, ELISPOT Chromium 51 CSFE proliferation assay Luciferase reporter TCR signaling assay | Cell lines, modified HLA expression (GANMO-1, MEG01, KAZZ, OUN-1) Primary AML samples CDX NOG mouse model |
| Spranger et al48 | HMMR (MSFPKAPL) | A∗02:01 | Healthy | Allo | RNA-loaded DCs | CD137 single-cell clones | PCR | Murine constant | - | RV | Healthy donor T cells | Chromium 51 CFU assay | Cell line, modified HLA expression (K562, THP1) Primary AML CDX NSG mouse model HSCs |
| MiHA | |||||||||||||
| Van Loenen et al49 | HA-1 (VLHDDLLEA) | A∗02:01 | Healthy | Not specified | Not specified | Not specified | Not specified | Cysteine modifications | - | RV | Allo-restricted healthy donor T cells | ELISA Cytotoxicity assays | EBV-LCLs Primary AML |
| Dossa et al50 | HA-1 (VLHDDLLEA) | A∗02:01 | Healthy | Auto | Peptide-loaded DCs | Single-cell clones | RACE PCR | Cysteine modifications | CD8 coreceptor iCasp9 safety switch | LV | Healthy donor T cells | Chromium 51 Intracellular cytokine release and CD107 degranulation flow cytometry | T2 peptide–loaded cells Primary AML |
| Pilunov et al51 | HA-1 | A∗02:01 | Healthy | Auto | Peptide-loaded DCs | CD137/multimter | RACE PCR | Murine constant Cysteine modifications | CRISPR-Cas9 knock down of endogenous TCR | LV | Healthy donor CD8+ cells | Fluorescent reporter J76 cell line assay ELISA Flow cytometry cytotoxicity assay | Primary AML cell lines, modified HLA expressed, peptide loaded (K562) Healthy and patient PBMCs |
| Neoantigens | |||||||||||||
| Van der Lee et al52 | mutNPM1 (CLAVEEVSL) | A∗02:01 | Healthy (unsuccessful with AML patients) | Auto | - | Tetramer single-cell clones | PCR | Murine constant Cysteine modification | - | RV | Healthy PBMCs, later CD4/8 purified | ELISA Chromium 51 | Peptide-loaded T2 cells Primary AML CDX NSG mouse model |
| Van der Lee et al53 | mutNPM1 (AVEEVSLRK) | A∗11:01 | Healthy | Auto | - | Tetramer single-cell clones | PCR | Murine constant | - | RV | Healthy PBMCs, later CD4/8 purified | ELISA Chromium 51 | Peptide-loaded T2 cells EBV-LCLs Cell lines, modified HLA expression (K562, OCI-AML2, OCI-AML3) Primary AML CDX NSG mouse model |
| Biernacki et al54 | CBFB-MYH11 fusion (REEMEVHEL) | B∗40:01 | Healthy | Auto | Peptide-loaded DCs | Single-cell clones | RACE PCR | Cysteine modifications | - | LV | Healthy donor T cells | Chromium 51 Flow cytometry–based cytotoxicity and CD107 degranulation assays | Peptide-loaded LCLs Cell lines, modified HLA/antigen expression (NB4, ME-1) PDX MISTRG mouse model |
| Biernacki et al55 | U2AF1Q157R (DFREACCRR) | A∗33:01/03 | Healthy | Auto | Peptide-loaded DCs | Single-cell clones | RACE PCR | Murine constant Cysteine modifications | CRISPR-Cas9 knock down of endogenous TCR | LV | Healthy donor T cells | Chromium 51 Flow cytometry–based cytotoxicity and CD107 degranulation assays | Peptide-loaded LCLs Cell lines, modified antigen expression (TF-1) Primary AML Healthy PBSCs/MCs CDX NSG mouse model |
| Giannakopoulou et al56 | mutFLT3 - D835Y (YIMSDSNYV) | A2 | Healthy | Auto | RNA-loaded DCs | Multimer single-cell clones | RT-PCR | Murine constant Cysteine modifications | - | RV | Healthy donor PBMCs | ELISA Flow cytometry–based activity and cytotoxicity assays | Cell lines, modified HLA expression, peptide-loaded (K562) EBV-LCLs Primary AML Healthy A2+ cells CDX and PDX NSG mouse model |
aa, amino acid; AAV, adeno-associated virus; B-CLL, B-cell chronic lymphocytic leukaemia; Cas9, CRISPR-associated protein 9; iCas9, inducible caspase 9; CDX, cell line-derived xenograft; CFU, colony-forming unit; CTLs, cytotoxic T lymphocytes; DCs, dendritic cells; EBV, Epstein-Barr virus; ELISA, enzyme-linked immunosorbent assay; ELISPOT, enzyme-linked immunospot; FACS, fluorescence-activated cell sorting; INFγ, interferon-γ; LCLs, lymphoblastoid cell lines; LDH, lactate dehydrogenase; LV, lentivirus; NSG, NOD scid gamma; NOD/SCID, nonobese diabetic/severe combined immunodeficiency; NOG, NOD/shi-scid IL2γr(null); PDX, patient-derived xenograft; PHA, phytohemagglutinin; RACE, rapid amplification of complementary DNA ends; RT, reverse transcription; RV, retrovirus; PCR, polymerase chain reaction.
Overexpressed antigens, known as leukemia-associated antigens can be used for generating broadly applicable TCR therapies. However, there is potential for on-target, off-tumor toxicities because of their expression on healthy cells. A subclass of overexpressed antigens known as cancer testes antigens have normal expression restricted to immune-privileged male gonadal tissue, making them particularly attractive targets with less risk of similar toxicities. Knowledge of antigen processing is essential because immunoproteasomes can be downregulated in AML, contributing to immune evasion. Peptides processed by alternative proteosomes were shown to be a more promising target.38
Minor histocompatibility antigens (MiHA) are a large class of 9 to 20 amino acid-long peptides presented on the HLA complex, often arising from single nucleotide polymorphisms or other normal variants, some with selective expression on hemopoietic cells. In AML, they are involved in mediating the therapeutic graft-versus-leukemia effect of allo-HSCT. MiHA antigen therapies can circumvent on-target, off-tumor toxicities when correct donors are MiHA mismatched. Although MiHA restriction and post–allo-HSCT setting may limit applicability, allo-HSCT remains a standard of care in AML. Current clinical trials targeting minor histocompatibility antigen HA-1 are still suitable for 10% to 15% of patients in the post-HCST setting.58
Targeting cancer-specific neoantigens in AML, which arise often because of splicing defects,59,60 can also avoid off-tumor toxicities.52-56 However, given the limited pool of patients possessing both the targeted mutation and the correct HLA, broad application of a specific singular neoantigen–directed product will be limited. Banks of TCR products targeting a wide range of neoantigens may need to be developed. Neoantigen-specific TCR T cells are promising for AML with the initiation of the first clinical trials (ClinicalTrials.gov identifier: NCT06424340) likely to provide insights into the feasibility and efficacy of this approach.61
Ultimately, targeting multiple antigens simultaneously will likely improve outcomes to cell therapies and can also help overcome acquired resistance because of the emergence of single antigen–escape variants.
TCR identification
PRAME-specific TCRs have been isolated from peripheral blood mononuclear cells (PBMCs) of patients44 that were not found in healthy donors41 and from CTLs generated from HLA-matched (autologous [auto]) or HLA-unmatched (allo) healthy donors36,37,52
Some have argued that for overexpressed antigens survivin, PRAME, or WT1, auto-HLA–restricted T cells have undergone negative thymic selection with the target antigen considered as “self,” and those donors would not have high affinity self-reactive or cross-reactive TCRs. TCRs from this pool theoretically should not be responsive to low levels of target antigen, which may be present in normal tissues, alleviating safety concerns for use. Indeed, a survivin-specific TCR identified from an allo-HLA–restricted donor was shown to exert fratricidal effects because of survivin expression on the T cells themselves,62 whereas another group was able to subsequently successfully identify a survivin TCR without the same effects from an auto-HLA–restricted donor.40
Conversely, others have also argued that the same thymic selection processes in allo-HLA–restricted donor PBMCs would allow for higher-avidity TCRs because these have not undergone the negative selection pressures for the targeted HLA. Amir et al44 have demonstrated that PRAME-specific allo-HLA–restricted T clones were of higher avidity than auto-HLA–restricted clone but also responded to healthy cell subsets, exemplifying some of the safety concerns associated with allo-HLA repertoire–derived TCRs. Falkenberg et al63 demonstrated similar results in which allo-HLA–restricted WT1 TCRs were more tumor reactive but also showed promiscuity against non-WT1 targets.
TCR identification typically involves coculturing donor PBMCs with antigen-presenting cells such as dendritic cells modified to overexpress the antigen of interest or directly loaded with antigenic peptides. Immunogenic epitopes can be predicted by algorithms such as NetMHC,64 or by testing peptide pools of overlapping 9-15mers spanning the length of the entire antigen.65,66 Activated, antigen-specific T cells are then isolated either using activation marker selection, most commonly CD137 or, if the target peptide/HLA is known, using major histocompatibility complex (MHC) multimers. Activated T-cell culture or CTL clones are expanded through repetitive stimulations with feeder cells.
Single-cell sequencing platforms or Sanger sequencing after rapid amplification of complementary DNA–ends polymerase chain reaction are used to precisely identify the α and β TCR subunits from the CTL cultures. Improved technologies with DNA barcoding of multimers allow for high throughput screening of multiple epitopes/HLAs.67 Alternatively, the inclusion of functional testing with high-throughput screening and sequencing pipelines can also shorten the developmental timeline.68,69
Transgenic TCR T-cell generation, optimization, and functional testing
Although the introduction of full-length sequences of the TCR α and β subunits into nonspecific T cells is sufficient to confer antigen specificity, recent exciting developments in lymphocyte engineering has allowed for modifications that will ensure precise pairing of the introduced subunits. The expressed subunits from these transgenes can associate with the endogenous CD3 subunits to form a functional multisubunit TCR.
Retroviral, lentiviral, or adeno-associated viral transduction are frequently used to introduce the α and β subunit sequences. More recently, nonviral methods such as CRISPR–associated protein 9 (CRISPR-cas9)–mediated insertion and the transposon Sleeping Beauty system have also been used.36,51,70,71 Such DNA-based methods are being investigated to increase the affordability of cellular therapies, with benefits of larger payloads and/or targeted integration.
Specific TCR modifications include replacement of the human constant region with a murine constant region, which does not interfere with TCR recognition and also allows for antibody-based detection of the introduced TCR in preclinical studies.72,73 Complementary cysteine (Cys) residues introduced into the constant region enable Cys-Cys disulphide bonds further ensuring specific pairing of the exogenous αß to prevent risk of mispairing with endogenous TCR αß chains.74,75 Mispairing could reduce antigen-specific activity to theoretically produce random new specificity with unpredictable toxicity. These design strategies highlight the potential advantages and evolving scope of synthetic immunology.
Antigen-specific TCR T cells are tested for functional response upon coculture with target cells by the release of effector cytokines such as interferon-γ and cytotoxicity assays. Often, TCR T cells are initially tested against peptide-loaded target cells to confirm specificity, and then against HLA-expressing cancer cell lines with either antigen overexpression or physiological antigen levels. A panel of HLA-expressing cell lines are used to rule out cross-reactivity.36 Testing against primary tumor tissue and in vivo murine models are often used for final confirmation before clinical translation. Although usually extensive targeted testing is carried out to confirm specificity after the TCR T-cell generation, recent studies have also used a large-scale approach with testing of T-cell clones before TCR sequencing to identify promising candidates from hundreds of clones.36,37 More recently, novel fluidics–based platforms such as the Bruker Beacon system are being developed for efficient, high-throughput TCR functional testing using single cells.76 This exciting technology can transform single-cell analysis because it has the advantage of screening thousands of cells for functional response, precisely capturing reactive cells for sequencing, and, thus, has the potential to revolutionize TCR identification and testing.
Affinity maturation of the TCR can be used to enhance efficacy. Amino acid substitutions in the CDR3a region of New York esophageal squamous cell carcinoma 1 (NY-ESO-1)–specific TCRs resulted in much greater reactivity against AML.77 Other methods include phage display78,79 and somatic hypermutation.80 Affinity-matured TCRs may require an additional focus on safety because an increased risk of toxicities has been seen in some studies. For instance, an affinity-matured TCR T-cell product targeting carcinoembryonic antigen led to transient severe colitis,81 and melanoma-associated antigens (MAGE)-A3–targeting TCRs with cross-reactivity to MAGE-A12 and titin82,83 resulted in neurotoxicity and cardiogenic shock in human trials. These cases demonstrate that although TCR modifications have their merit, extensive safety considerations are also required. However, Afamicel, an affinity-matured TCR T-cell product targeting MAGE-A4, was recently US Food and Drug Administration approved for synovial sarcoma, paving the way for other TCRs to be approved in the future.
Cross-reactivity of the TCRs can be investigated using structural and predictive algorithms. Single amino acid substitutions using alanine (alanine scans) or other amino acids (X-scans) can be used for evaluating potential peptides and position of key amino acids that interact with the TCR.84,85 Combinatorial peptide libraries can also be used to test the TCR reactivity using in vitro assays, including cytokine release or target cell lysis. Although computational modeling to predict peptide-MHC binding such as EPIC-TRACE,86 and ERGO (peptide-TCR matching prediction)87,88 are being developed, the precise rules for TCR-peptide binding are still not well established.23 Empiric testing involving large peptide libraries encoded in yeast or baculoviral expression systems or using PresentER in which peptides can be loaded on to endogenous MHC of mammalian cells through endoplasmic reticulum signaling sequences for in vitro and in vivo testing are being developed. Other library screening techniques described include SABR (signaling and antigen-presenting bifunctional receptors) and T-scan reporter systems, which mimic endogenous presentation.89 Testing for off-target peptide binding is crucial to establish safety before clinical translation and we envision several new algorithms being described in the near future.
Novel designs
Advances in molecular biology and genetic engineering have seen the development of exciting novel receptors, that have extended the original chimeric designs described by Gross et al in 1989.90 Some of these designs are likely to dictate the success of cell therapies. The reader is directed to Table 3 in which additional features and novel chimeras are highlighted.
Novel designs to enhance TCR T-cell functional efficacy and safety, and overcome HLA-restriction limitations

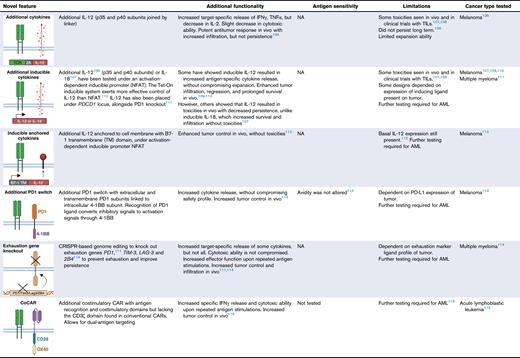

ATAM, artificial T-cell adaptor molecule; CML, chronic myeloid leukemia; CMV, cytomegalovirus; GVHD, graft-versus-host disease; LAG-3, lymphocyte activayion gene 3; NA, not applicable; NFAT, nuclear factor of activation T cells; PD1, programmed cell death 1; PDCD1, programmed cell death protein 1; PD-L1, programmed death ligand 1; TILs, tumor-infiltrating lymphocytes; TIM-3, T-cell immunoglobulin and mucin-domain containing 3; TNFα, tumor necrosis factor α; siRNA, small interfering RNA; STAR, synthetic TCR and antigen receptor; TACs, T-cell antigen couplers; tEGFR, truncated epidermal growth factor receptor; TRAC, T-cell receptor α constant; TRBC, T-cell receptor β constant; TRuCs, T-cell receptor fusion constructs. Figures created with BioRender.com: Gore S. (2025); https://BioRender.com/o91f055
Improving functional efficacy
Expression of coreceptors and costimulatory molecules
TCR activation relies on the association of coreceptors and signaling domains (Figure 1). The insertion of such additional CD3 subunits,97 CD8 receptor,50,91,92,94,95 and 4-1BB costimulatory molecules98,133 was a logical approach to increase signaling efficacy and to boost TCR T-cell potency.
Alternatively, new designs have investigated linking the TCR αβ chains to downstream intracellular signaling domains of CD3ζ, analogous to that of a CAR,100 or to CD28-CD3ε costimulatory domains.103 Similarly, the 4-1BB costimulatory molecule inserted into the CD3ζ, termed an artificial T-cell adaptor molecule was shown to have a higher proliferative capacity when highly expressed alongside the NY-ESO-1 TCR.104,105 These represent viable avenues for improving TCR T-cell efficacy, with particular application for low-avidity TCRs with a favorable safety profile but lacking the required potency.
Enhancing cytokine-mediated activation
Immune modulating cytokines exert either stimulatory or suppressive signaling and inclusion of transgenes encoding for cytokines within the T cells, overcomes the requirement for exogenous infusions. For example, transgenes encoding interleukin-12 (IL-12), T-cell stimulatory cytokine,106 can be placed under an activation inducible nuclear factor of activated T cells promoter109 or a doxycycline-inducible Tet-On system, for efficient secretion from the modified T cells.110 Alternative approaches involve anchoring the IL-12 to the cell membrane, or the TCR itself, for a targeted approach, which showed efficacy in remodeling the TME in solid cancers.112 IL-18 expression demonstrated similar efficacy as IL-12, without the toxicity seen in in vivo models.107 These studies have been conducted for solid tumors and use in the context of AML may require further investigation because of the unique microenvironment of hematological malignancies.
Reversing immune suppression
AML cells can upregulate immune checkpoint ligands to induce T-cell exhaustion, limiting TCR T-cell efficacy and contributing to AML relapse. Switch receptors have been designed to convert the immune checkpoint inhibitory signals into activation signals.92,134,135 Most commonly, programmed cell death protein 1 immune checkpoint can be linked to an intracellular 4-1BB activation domain, facilitating an activation signal instead of the usual negative signal.113 CRISPR-mediated programmed cell death protein 1 knockout has also been combined with an IL-2 knockin to abrogate exhaustion and boost efficacy.111 Knock out of other exhaustion related genes such as T-cell immunoglobulin and mucin domain 3, lymphocyte activation gene 3, and 2B4 (also known as cluster of differentiation 244) results in improved persistence and response upon rechallenge.114 Such approaches have shown promise when paired with CAR T cells in early-phase clinical trials,136 with potential for similar application in TCR T cells.
Dual-antigen targeting
A well-documented driver of relapse in AML is the expansion of clones with downregulated target antigen expression. Dual-antigen–targeting strategies are being rapidly developed. Increasing precision targeting can be achieved by costimulatory CAR designs coexpressing an antigen-specific single-chain variable fragment (scFv) with a costimulatory domain but lacking the CD3ζ signaling. A surviving-targeting TCR, combined with such a scFv + costimulatory combination showed efficacy, without excessive CD3 activation.115 Alternatively, dual-expressing TCR CAR T cells have shown functional coefficacy in other malignancies, with initial studies showing promise for application in AML.116 Further studies will be required to confirm whether such dual-targeting approaches are successful in improving responses and overcoming therapy resistance induced by emergence of antigen-escape variants.
Addressing safety concerns
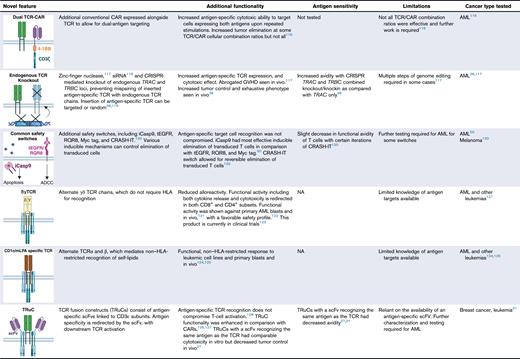
Removal of endogenous TCR
One potential safety risk is mispairing of the introduced TCR αβ with endogenous TCR αβ chains, resulting in unexpected specificity and toxicity. The deletion of endogenous TCR using zinc-finger nucleases,117 or knockout using small interfering RNA or CRISPR-CRISPR–associated protein 9 as shown in WT1- and NY-ESO-1–directed TCR T cells for AML, can abrogate the risk.36,118,119 The CRISPR-mediated knock out can be accompanied by the insertion of the antigen-specific TCR into the natural TRAC and TRBC locus, not only limiting risk of mispairing but also preventing excessive activation because the antigen-specific TCR is now expressed at physiological levels under endogenous promoter controls.137,138
Safety switches for unexpected toxicities
In the event of unexpected toxicity, safety switches to induce apoptosis of the infused T-cell product to abrogate further deleterious effects have been designed. Commonly used is the inducible caspase 9, with which infusion of a dimerizing agent allows for controllable dimerization of caspase 9 to activate the downstream apoptosis pathway.139 Others include truncated epidermal growth factor receptor, which causes antibody-dependent cellular cytotoxicity upon interaction with cetuximab antibody,140 a chimeric molecule called RQR8 comprising CD20/CD34 epitopes to be eliminated with ritxuximab via antibody-dependent cellular cytotoxicity,141 and Myc-tag.142 Each of these safety switches was coexpressed with HA-1 TCR, with inducible caspase 9 the most effective in eliminating TCR T cells.50 More recently, the “CRASH-IT” switch explores a reversible safety switch wherein the addition of various drugs resulted in a dose-dependent, reversible proteasomal degradation of the TCR.120
Overcoming HLA restriction
Downregulation of the HLA molecule on AML blasts results in immune evasion, because TCR T cells are dependent on HLA presentation of targets.
HLA-independent TCRs
γδ T cells that make up ∼10% of T cells recognize cancer-specific phosphoantigens, in a HLA-independent manner.143 γδ TCRs can be inserted into αβ T cells for antigen recognition and cytotoxic response without HLA restriction.121 Because the γδ TCRs do not pair with αβ chains, any risk of mispairing-mediated toxicity is removed. One such product, TEG1001, is moving into early-phase clinical trials.122
Some alternate natural TCRs can recognize HLA-independent lipid molecules such as CD1c.124,144 One such TCR recognizing methyl-lysophosphatidic acid, which is selectively upregulated on AML cells, was shown to delay AML progression in xenograft mice models.125 Such a product allows for a broader use in patients but is also greatly limited by the type of antigens that can be targeted.
Antibody-based antigen recognition
Other novel designs have combined antibody-based antigen recognition, with downstream TCR clustering and signaling. scFvs have been linked to CD3ε subunits (TRuCs),126,127 or to an anti-CD3 antibody and CD4 transmembrane domain (T-cell antigen couplers) to overcome HLA restriction while maintaining a sensitive functional response.128 Alternatively, separate light and heavy chains can be attached independently to TCR α and β chain constant domains. These formats, named synthetic TCR and antigen receptors129 or HLA-independent TCRs130 have shown equivalent or greater responsiveness and tumor control over TCR formats, as well as TRuCs.27,31 In a similar concept, antibody TCRs link the separate antibody chains to TCR γ and δ chains.132 These designs combine the superior sensitivity of TCR signaling with conventional CAR-like antigen recognition.
Furthermore, CAR-like designs centered around TCR-like antibodies can recognize peptide-MHC complexes. Such peptide-centric CARs145 and TCR-mimic CARs146 have shown promise for AML but are still HLA restricted and use CAR-like downstream signaling. Direct comparisons indicate that TCR-based designs convey superior sensitivity and functionality in low –antigen density contexts over CARs, which are ideal for AML neoantigens.27,31
These technologies hold great promise in contributing to further detailed understanding of TCR and CAR signaling mechanisms, which will lead to fine-tuning of customized designs for the AML context.
Clinical trials with TCR T cells
The clinical trials of antigen-specific TCR T-cell therapy for AML are summarized in Table 4. WT1 was the target in 6 of 12 trials, with other trials targeting PRAME (2/12), HA-1/HA-2 (3/12), or mutated nucleophosmin 1 (dNPM1) (1/12). The source of production of TCR T-cell products were either autologous (n = 8) or allogeneic (n = 4), and 11 studies targeted HLA-A∗02:01–restricted antigens.
TCR T-cell therapy for clinical trials for AML
| Trial reference | Phase/status | Product details | Disease | Disease type | Patients treated | Response | Persistence | Significant adverse events | |||
|---|---|---|---|---|---|---|---|---|---|---|---|
| UMIN000011519147 PMID: 28860210 | Phase 1 Completed | Retroviral vector with siRNA knock down of endogenous TCR, WT1 peptide vaccine | Auto | WT1 | A∗24:02 | Refractory AML, MDS | Active | 8 (2 AML) | 2/8 showed decreased blast counts in the BM (predicted leukemia regression). 2 patients with AML did not respond. | Yes, at 5 mo after treatment (4/5 patients survived for ≥12 mo). | No serious adverse events reported. |
| NCT01621724 EudraCT-2006-004950-25 | Phase 1/2 Completed | Retroviral vector, IL-2 standard conditioning | Auto | WT1 | A∗02:01 | AML, CML | Active | 7 | 4 patients showed disease responsiveness. No response in 3 patients. | Yes, for 4/7 patients at 1 y after treatment. | Febrile neutropenia (1). Anemia (1). |
| NCT01640301148 PMID: 31235963 | Phase 1/2 Terminated | EBV-specific CD8+ T cells, Additional IL-2 injection | Allo | WT1 | A∗02:01 | AML (recurrent/secondary) having undergone allo-HSCT (with no evidence of disease) | In remission | 12 | Maintenance of remission at median of 44 mo for 12 patients. | Yes, for 4/12 patients at 1 y after treatment. | CRS grade 3 (2). Neutropenia (2). Thrombocytopenia (2). Lymphopenia (12). Anemia (7). |
| NCT02550535115 EudraCT-2014-003111-10 | Phase 1/2 Completed | Retroviral vector, Additional IL-2 injection | Auto | WT1 | A∗02:01 | AML, MDS | In remission | 10 (AML) | Median survival of 12 mo in 6 patients with AML. | Yes, for 7/10 patients over 12 mo. | CRS (1). |
| NCT05066165 | Phase 1/2 Terminated | CRISPR/Cas9 | Auto | WT1 | A∗02:01 | AML | Active | 2 | Disease progression in both patients. | Not reported. | Febrile neutropenia (1). |
| NCT02770820 | Phase 1/2 Terminated | EBV-specific CD8+ TCM/TN T cells, Additional IL-n2 injection | Auto | WT1 | A∗02:01 | High-risk non-M3 AML (with prior consolidation chemotherapy) | In remission | 7 | Not reported. | Not reported. | No serious adverse events reported. |
| NCT03503968149 EudraCT-2017-000440-18 | Phase 1/2 Terminated | - | Auto | PRAME | A∗02:01 | AML, MDS, MM | Active | 9 | No disease progression for 1 patient. Remission at 4 wk followed by progression at 3 mo for 1 patient. Disease progression in remaining patients (7/9). | Yes, for 6/8 patients at 4 wk. | CRS grades 1-2 (2). Other SAE (not specified) (5). |
| NCT02743611 | Phase 2/2 Terminated | Includes safety switch activated with rimiducid | Auto | PRAME | A∗02:01 | Relapsed AML, MDS, uveal melanoma | Active | 4 | Not reported. | Not reported. | Neutropenic fever, tachypnea, CRS, pseudomonas bacteremia infection, neurotoxicity, orthostatic hypotension (1). |
| NCT03326921150 PMID: 38683966 | Phase 1 Suspended | CD8+ CD4+ TM T cells | Allo | HA-1 HA-1 (H) genotype (RS_1801284: A/G, A/A) | A∗02:01 | Pediatric and adult leukemias after allo-HCT | Active/in remission | 9 | Reduction of marrow blasts lasting >30 days (2/9). Sustained remission (2/9). Disease progression in remaining patients (5/9). | Yes, for 8/9 patients beyond 1 y after treatment. | Neutropenia (5). Fever (3). Infection (2). Infusion reaction (1). |
| EudraCT-2010-024625-20151 PMID: 32973756 | Phase 1 Terminated | EBV- or CMV-specific T cells retroviral vector | Allo | HA-1 | A∗02:01 | High-risk leukemia, after allo-HCT | Active (1)/in remission (4) | 5 | Maintenance of relapse-free survival at follow-up in 2 patients. Disease progression in 3 patients. | Yes, for 2 patients up to 21 wk. | Neutropenia. Thrombocytopenia (1). |
| NCT05473910152,153 | Phase 1 Recruiting | - | Allo | HA-1 or HA-2 | A∗02:01 | AML, MDS, ALL undergoing haplo-identical allo-HCT | In remission | 7 | Maintenance of remission at a median follow-up of 162 d in all patients (at time of reporting). | Yes, ongoing persistence at longest follow-up of 203 d. | GVHD (4). |
| NCT06424340 | Phase I/II Recruiting | - | Auto | dNPM1 | A∗02:01 | AML (relapsed or refractory) | Active | - | Study ongoing | Study ongoing | Study ongoing |
| Trial reference | Phase/status | Product details | Disease | Disease type | Patients treated | Response | Persistence | Significant adverse events | |||
|---|---|---|---|---|---|---|---|---|---|---|---|
| UMIN000011519147 PMID: 28860210 | Phase 1 Completed | Retroviral vector with siRNA knock down of endogenous TCR, WT1 peptide vaccine | Auto | WT1 | A∗24:02 | Refractory AML, MDS | Active | 8 (2 AML) | 2/8 showed decreased blast counts in the BM (predicted leukemia regression). 2 patients with AML did not respond. | Yes, at 5 mo after treatment (4/5 patients survived for ≥12 mo). | No serious adverse events reported. |
| NCT01621724 EudraCT-2006-004950-25 | Phase 1/2 Completed | Retroviral vector, IL-2 standard conditioning | Auto | WT1 | A∗02:01 | AML, CML | Active | 7 | 4 patients showed disease responsiveness. No response in 3 patients. | Yes, for 4/7 patients at 1 y after treatment. | Febrile neutropenia (1). Anemia (1). |
| NCT01640301148 PMID: 31235963 | Phase 1/2 Terminated | EBV-specific CD8+ T cells, Additional IL-2 injection | Allo | WT1 | A∗02:01 | AML (recurrent/secondary) having undergone allo-HSCT (with no evidence of disease) | In remission | 12 | Maintenance of remission at median of 44 mo for 12 patients. | Yes, for 4/12 patients at 1 y after treatment. | CRS grade 3 (2). Neutropenia (2). Thrombocytopenia (2). Lymphopenia (12). Anemia (7). |
| NCT02550535115 EudraCT-2014-003111-10 | Phase 1/2 Completed | Retroviral vector, Additional IL-2 injection | Auto | WT1 | A∗02:01 | AML, MDS | In remission | 10 (AML) | Median survival of 12 mo in 6 patients with AML. | Yes, for 7/10 patients over 12 mo. | CRS (1). |
| NCT05066165 | Phase 1/2 Terminated | CRISPR/Cas9 | Auto | WT1 | A∗02:01 | AML | Active | 2 | Disease progression in both patients. | Not reported. | Febrile neutropenia (1). |
| NCT02770820 | Phase 1/2 Terminated | EBV-specific CD8+ TCM/TN T cells, Additional IL-n2 injection | Auto | WT1 | A∗02:01 | High-risk non-M3 AML (with prior consolidation chemotherapy) | In remission | 7 | Not reported. | Not reported. | No serious adverse events reported. |
| NCT03503968149 EudraCT-2017-000440-18 | Phase 1/2 Terminated | - | Auto | PRAME | A∗02:01 | AML, MDS, MM | Active | 9 | No disease progression for 1 patient. Remission at 4 wk followed by progression at 3 mo for 1 patient. Disease progression in remaining patients (7/9). | Yes, for 6/8 patients at 4 wk. | CRS grades 1-2 (2). Other SAE (not specified) (5). |
| NCT02743611 | Phase 2/2 Terminated | Includes safety switch activated with rimiducid | Auto | PRAME | A∗02:01 | Relapsed AML, MDS, uveal melanoma | Active | 4 | Not reported. | Not reported. | Neutropenic fever, tachypnea, CRS, pseudomonas bacteremia infection, neurotoxicity, orthostatic hypotension (1). |
| NCT03326921150 PMID: 38683966 | Phase 1 Suspended | CD8+ CD4+ TM T cells | Allo | HA-1 HA-1 (H) genotype (RS_1801284: A/G, A/A) | A∗02:01 | Pediatric and adult leukemias after allo-HCT | Active/in remission | 9 | Reduction of marrow blasts lasting >30 days (2/9). Sustained remission (2/9). Disease progression in remaining patients (5/9). | Yes, for 8/9 patients beyond 1 y after treatment. | Neutropenia (5). Fever (3). Infection (2). Infusion reaction (1). |
| EudraCT-2010-024625-20151 PMID: 32973756 | Phase 1 Terminated | EBV- or CMV-specific T cells retroviral vector | Allo | HA-1 | A∗02:01 | High-risk leukemia, after allo-HCT | Active (1)/in remission (4) | 5 | Maintenance of relapse-free survival at follow-up in 2 patients. Disease progression in 3 patients. | Yes, for 2 patients up to 21 wk. | Neutropenia. Thrombocytopenia (1). |
| NCT05473910152,153 | Phase 1 Recruiting | - | Allo | HA-1 or HA-2 | A∗02:01 | AML, MDS, ALL undergoing haplo-identical allo-HCT | In remission | 7 | Maintenance of remission at a median follow-up of 162 d in all patients (at time of reporting). | Yes, ongoing persistence at longest follow-up of 203 d. | GVHD (4). |
| NCT06424340 | Phase I/II Recruiting | - | Auto | dNPM1 | A∗02:01 | AML (relapsed or refractory) | Active | - | Study ongoing | Study ongoing | Study ongoing |
BM, bone marrow, CRS, cytokine release syndrome; MDS, myelodysplastic syndrome; MM, myelodysplastic syndrome; SAE, severe adverse event; siRNA, small interfering RNA; TCM, central memory T cell; TM, memory T cell; TN, naive T cell.
Following from promising WT1-directed CTLs, in trial NCT01640301, 12 patients with AML were treated prophylactically with allo-HCT donor–derived Epstein-Barr virus–specific CD8+ WT1 TCR T cells after allo-HSCT. All patients achieved relapse-free survival at a median follow-up of 44 months. In a comparative cohort, there was a relapse-free survival rate of only 54%, indicating that the treatment likely has some efficacy in preventing relapse.148 Trial NCT01621724 treated 7 patients with AML or chronic myeloid leukemia, disease status not specified, with autologous WT1–directed TCR T cells, 4 of whom showed disease responsiveness.154 A similar product was used in NCT02550535, in which all 6 prophylactically-treated patients with AML remained in remission at follow-up (median of 12 months).149 In these 3 trials, persistence of T cells was shown in 33% to 70% of patients at 1 year. Generally, the treatment was well tolerated, with no on-target, off-tumor toxicities. Adverse events included cytokine release syndrome, and, some instances of neutropenia, anemia, thrombocytopenia, and lymphopenia that resolved in all cases.
It is important to note that in 2 of the aforementioned trials, patients enrolled were in remission at the time of the treatment. Thus, direct contribution of the TCR T cells in preventing relapse is not clear. Indeed, 1 trial treated 8 patients with AML or refractory myelodysplastic syndrome with an autologous WT1 TCR T cell, which had small interfering RNA–mediated endogenous TCR knockdown and an additional WT1 peptide vaccine treatment (UMIN000011519). Of 8 patients, 2 showed decreased blast counts the remaining 6 patients did not respond (including 2 patients with AML). Still, T cells persisted for 5 months in 5 of patients (of whom 4 survived past 12 months), and no treatment-related toxicities or adverse events were detected.147 Similarly, 2 patients with AML with detectable disease were treated in trial NCT05066165, with both patients experiencing disease progression without severe adverse events after treatment. These 2 trials spoke to the safety of TCR T-cell therapies, and indicated their potential for preventing relapse, which is a significant challenge in AML. Undeniably, further study is required in this area.
There have also been 2 TCR T-cell trials with a small number of participants (ClinicalTrials.gov identifier: NCT03503968 and NCT02743611) targeting the cancer testes antigen HLA -A∗02:01/PRAME with AML, myelodysplastic syndrome, and uveal melanoma, which have been completed and results awaited. In addition, a recent trial has been initiated targeting a neomutation in dNPM1 (ClinicalTrials.gov identifier: NCT06424340).
Three phase 1 clinical trials for patients with AML or other leukemias, undergoing allo-HSCT have been initiated using HA-1–specific T cells. Although 1 trial is still open (ClinicalTrials.gov identifier: NCT05473910155), results for the EudraCT-2010-024625-20 and NCT03326921 trials have been reported. In the former, 5 patients positive for HA-1 were treated prophylactically with Epstein-Barr virus– or cytomegalovirus-specific CD8+ HA-1 TCR T cells generated from allo-HA-1–negative donors.151 Of these, 2 patients remained in remission throughout the study duration and HA-1 T cells persisted but did not appear to expand in vivo. No graft-versus-host disease or toxicity was observed. Trial NCT03326921, a CD8 and CD4 TCR T-cell product incorporating a CD8 coreceptor with the HA-1 TCR, treated 9 patients who had relapsed early after allo-HSCT, some of whom had achieved another remission. T cells persisted in 8 patients for up to a year, and disease responsiveness was seen in 4 patients, with 1 maintaining complete remission for >27 months.
This is a rapidly evolving field, and the handful of trials completed thus far have provided key insights. The paucity of severe adverse events seen in most trials, and sustained remission accompanied by long-term persistence of the therapeutic T cells, even with low doses, seen in some patients has been very promising. One common limitation resulting in early termination of trials was slow accrual, likely because of the strict HLA and antigen-specificity requirements for this sort of therapy. Furthermore, inability to generate products for all enrolled patients is another limitation. In the EudraCT-2010-024625-20 trial almost half (4/9 patients) were not treated for this reason. In autologous programs, this is made more difficult because patient T-cell quality may be affected by previous chemotherapy and/or other treatments. Indeed, in the NCT05066165 trial, only 2 patients were treated before the trial was terminated to move to an allogeneic version of the same WT1 TCR therapy. Thus, larger trials with increased patient recruitment are required to be able to draw more reliable conclusions. Platform trials using TCR T cells to multiple antigens, HLAs, and/or multiple cancers may be an option. Upcoming phase 2 trials will provide further information regarding the efficacy of these products for AML.
Conclusion
TCR therapy is poised to revolutionize treatment for AML as evidenced by the substantial preclinical development of TCR T cells and promising early-phase clinical trials. Although it is premature to predict which 1 of the developed options will become an approved treatment, the favorable safety profiles encourage further testing. Novel designs to enhance efficacy and safety are being fine-tuned. It is expected that combining engineering strategies with high throughput identification and testing, along with the development of algorithms and in silico analyses will facilitate rapid development of TCR therapies. The technologies will also likely enable improving responses and reducing off-target toxicities. Thus, TCR therapy is expected to become a crucial component in the T-cell therapeutic arsenal for AML treatment, and given AML’s complex immunophenotype, combinatorial targeting of multiple antigens will likely be necessary.
Acknowledgments
The authors are grateful for critical review and comments from Geraldine O’Neill, Head of Children’s Cancer Research Unit, The Children’s Hospital at Westmead, and The University of Sydney.
K.G. reports salary support from the University of Sydney. S.G. reports a Research Training Program Scholarship (Government of Australia) and support for research in the laboratory: CCRU Department (CHW amd Dooley's Foundation, Lidcombe, Australia).
Authorship
Contribution: K.G. conceived and drafted the manuscript outline; S.G. wrote the manuscript and generated the figures with input, reviews, and edits from K.G.; E.B. and M.B. read the manuscript and provided critical feedback and additional key information; K.L. and K.M. reviewed and provided additional information on clinical trials; and S.G. and K.G. researched the topic and primarily focused on the preclinical development, antigens, and novel designs whereas the clinician researchers reviewed “Clinical trials with TCR T-cells.”
Conflict-of-interest disclosure: M.B. is an inventor on a patent describing HA-1 TCR T cells that was previously licensed to Elevate Bio and has recently been licensed to Promicell Inc; received research funding from HighPass Bio, an Elevate Bio portfolio company; and has financial interests in HighPass Bio and Promicell Inc. E.B. and K.M. hold patents in adoptive cell therapy for opportunistic infection and malignancy. E.B. reports advisory board membership for IQVIA, AbbVie, MSD, Astellas, Novartis, Gilead, and Bristol Myers Squibb, and research funding from MSD. The remaining authors declare no competing financial interests.
Correspondence: Kavitha Gowrishankar, The University of Sydney, CCRU, CHW, 178 Hawkesbury Rd, Westmead, Sydney, NSW 2145, Australia; email: kavitha.gowrishankar@sydney.edu.au.

