

Key Points
The p.(Cys1084Tyr) variant causes a variably expressed, complex, mixed phenotype form of von Willebrand disease.
p.(Cys1084Tyr) VWF differentially associates with qualitative and/or quantitative defects in heterozygous and homozygous individuals.

Abstract
von Willebrand factor (VWF) is an extremely cysteine-rich multimeric protein that is essential for maintaining normal hemostasis. The cysteine residues of VWF monomers form intra- and intermolecular disulfide bonds that regulate its structural conformation, multimer distribution, and ultimately its hemostatic activity. In this study, we investigated and characterized the molecular and pathogenic mechanisms through which a novel cysteine variant p.(Cys1084Tyr) causes an unusual, mixed phenotype form of von Willebrand disease (VWD). Phenotypic data including bleeding scores, laboratory values, VWF multimer distribution, and desmopressin response kinetics were investigated in 5 members (2 parents and 3 daughters) of a consanguineous family. VWF synthesis and secretion were also assessed in a heterologous expression system and in a transient transgenic mouse model. Heterozygosity for p.(Cys1084Tyr) was associated with variable expressivity of qualitative VWF defects. Heterozygous individuals had reduced VWF:GPIbM (<0.40 IU/mL) and VWF:CB (<0.35 IU/mL), as well as relative reductions in high-molecular-weight multimers, consistent with type 2A VWD. In addition to these qualitative defects, homozygous individuals also displayed reduced factor VIII (FVIII):C/VWF:Ag, leading to very low FVIII levels (0.03-0.1 IU/mL) and reduced VWF:Ag (<0.40 IU/mL) and VWF:GPIbM (<0.30 IU/mL). Accelerated VWF clearance and impaired VWF secretion contributed to the fully expressed homozygous phenotype with impaired secretion arising because of disordered disulfide connectivity.
Introduction
The basic von Willebrand factor (VWF) functional unit, the monomer, exhibits considerable internal homology and is composed of a series of repeated domains stabilized by extensive intramolecular disulfide bonding.1 Highly conserved cysteine residues are dispersed throughout the VWF sequence but cluster mainly in the N and C termini, where they participate in intermolecular disulfide interactions that are essential for the formation of high-molecular-weight multimers (HMWM). VWF monomers become disulfide-linked at the C terminus within the endoplasmic reticulum (ER) to form VWF dimers that are in turn disulfide linked to form HMWM as they pass through the Golgi apparatus and are packaged into Weibel Palade bodies (WPBs) for secretion. Previously, it was suggested that initial pairing of all cysteines is critical to ensure correct VWF folding during the initial stages of synthesis. However, a proportion of plasma-derived and recombinant VWF molecules have been reported to contain unpaired cysteine residues.2-5
Cysteine residues and their associated disulfide bonds are critical for maintaining VWF structure and function, and the functional roles of several VWF disulfide bonds have been defined. In particular, long-range intramolecular disulfides in the VWF A1 and A3 domains, as well as a rare vicinal disulfide between p.(Cys1669) and p.(Cys1670) in the A2 domain, play key roles in regulating the hemostatic activity and proteolytic processing of VWF.6-8
Cysteine variants account for ∼5% of all pathogenic VWF variants and are typically associated with highly penetrant forms of von Willebrand disease (VWD).9-11 Interestingly, accumulating evidence suggests that cysteine variants are associated with complex VWD phenotypes. For example, well-characterized cysteine variants p.(Cys1130Phe) and p.(Cys1149Arg) exert dominant-negative effects on VWF secretion, cause accelerated VWF clearance, and lead to aberrant multimerization resulting in overlapping type 1/type 2A VWD phenotypes.12-15
In this study, we report and characterize a previously undocumented cysteine variant, p.(Cys1084Tyr), that causes a complex, mixed phenotype VWD. We show that loss of this critical cysteine residue differentially associates with qualitative and/or quantitative VWF defects in heterozygous and homozygous individuals causing a variably expressed form of VWD.
Methods
Study approval
Study approval was obtained from the Research Ethics Board of the Hospital for Sick Children, Toronto, Canada, and all study participants gave written informed consent.
Bleeding questionnaires
Bleeding scores were evaluated by the International Society on Thrombosis and Hemostasis Bleeding Assessment Tool for adult family members and the Pediatric Bleeding Questionnaire for pediatric subjects by an expert administrator.16,17
DNA sequencing
EDTA-anticoagulated whole blood was obtained from all family members, and genomic DNA was isolated from leukocytes using the Gentra Puregene extraction kit (Qiagen, Germantown, MD, USA). The entire coding region of VWF and F8 of the index case (IC) was sequenced by Sanger methodologies and intron/exon boundaries, the 5′ and 3′ untranslated regions, and the proximal promoter regions of F8. The presence of the novel variant was confirmed via bi-directional sequencing in the IC. Direct sequencing of VWF exon 25 was carried out to detect the variant in the additional family members. Sequencing data can be found in GenBank (ncbi.nlm.nih.gov/genbank, accession numbers OM169364, OM169365, and OM169366).
Coagulation studies
VWF:Ag was determined by enzyme-linked immunosorbent assay (ELISA) using polyclonal VWF antibodies A0082 and P0226 (Dako, Glostrup, Denmark). VWF:GPIbM was measured using the INNOVANCE VWF Ac Assay (Siemens Healthcare Diagnostics, Marburg, Germany), and collagen-binding activity was determined as before.18,19 VWF propeptide was measured via an in-house ELISA using monoclonal capture antibodies 239.2 and 239.3 and biotinylated 242.2 and 242.6 for detection. VWF propeptide antibodies were provided by S. Haberichter (Versiti, Blood Center of Wisconsin, Milwaukee, WI). Factor VIII (FVIII):C was measured by 1-stage activated partial thromboplastin time clotting assay using an automated coagulometer (Siemens BCS XP, Oakville, ON, Canada).
Desmopressin challenge
Desmopressin (0.3 μg/kg) was administered IV, and blood was drawn at baseline and 1, 3, and 6 hours after administration. The VWF:Ag, VWF:GPIbM, and FVIII:C responses were plotted as concentration against time, and data were fitted to a mono-exponential curve. The 1-hour time point was taken as the beginning of the elimination phase. Half-lives were calculated from the first-order rate constant (k) for the elimination phase.
Multimer analysis
VWF multimers were analyzed as previously described.20
Expression plasmids and mutagenesis
The VWF expression vectors, pCIneohuVWF for recombinant human VWF protein production, and pSC11ETmVWF for hydrodynamic delivery have previously been described.20 Expression plasmid pCIneohuVWF was used as a template to generate the p.(Cys1084Tyr), p.(Cys1060Ala), and p.(Cys1060Ser) expression plasmids via site-directed mutagenesis using the Q5 Site-Directed Mutagenesis Kit (New England Biolabs, Whitby, ON, Canada). Expression plasmid pCIneohuVWF p.(Cys1084Tyr) was subsequently used as a template to generate p.(Cys1060Ala;Cys1084Tyr) and p.(Cys1060Ser;Cys1084Tyr). Site-directed mutagenesis was also performed on the pSC11ETmVWF expression vector to generate pSC11ETmVWF p.(Cys1084Tyr).
Protein expression studies
HEK 293T cells were transiently transfected with variant or wild-type (WT) VWF cDNA via liposomal transfer according to the manufacturer’s instructions (Lipofectamine 2000, ThermoFisher Scientific, Carlsbad, CA). Following transfection, cells were grown for 24 hours in Dulbecco’s modified Eagle’s medium (Life Technologies, Grand Island, NY) with 10% (vol/vol) fetal bovine serum (Wisent Bioproducts, Saint-Jean-Baptiste, QC, Canada) and for a further 48 hours in OptiMEM serum-free medium (Life Technologies, Grand Island, NY). Conditioned media were collected, and secreted VWF was concentrated using Amicon Ultra 0.5-mL 50K molecular weight cut-off centrifugal filter devices (Millipore, Cork, Ireland). VWF:Ag was measured in conditioned medium and in cell lysates by VWF:Ag ELISA.
Animal studies
Animal experiments were approved by the Queen's University Animal Care Committee (Kingston, ON, Canada). VWF−/− mice on a C57Bl/6 background (8-12 weeks) were used. For clearance studies, VWF was administered at 7.5 µg/mouse by tail vein injection. In vivo VWF expression was induced via hydrodynamic injection of Vwf cDNA as previously described.21
Immunocytochemistry
Immunocytochemical analysis of transiently transfected HEK 293 was carried out as described previously.22 Imaging was performed using a Leica TCS SP8 confocal microscope (Leica Microsystems, Concord, ON, Canada). Individual cells were imaged through a z-stack (Z-dimension of 2 to 3 μm) acquisition mode using the HC PL APO CS2 63×/1.40 oil objective. Images were deconvolved using Huygens Essential Software (Scientific Volume Imaging, Hilversum, Netherlands) and analyzed using LAS-X Software (Leica Microsystems, Concord, ON, Canada).
Immunohistochemistry
Diaminobenzidine (DAB) peroxidase staining for VWF expression by mouse hepatocytes following hydrodynamic injection was performed on formalin-fixed paraffin-embedded tissue using a rabbit polyclonal antibody (A0082; DAKO, Glostrup, Denmark) on a Ventana Discovery Immunostainer (Ventana Medical System, Tucson, AZ, USA).
Results
Identification of a novel cysteine variant in a consanguineous family with VWD
The IC (III-2) is a 9-year-old girl diagnosed with VWD following episodes of epistaxis requiring medical intervention. Sequencing of the VWF gene revealed a G to A transition (c.3251G>A) in exon 25, predicting a cysteine to tyrosine substitution at amino acid 1084 in the C8-3 subdomain of the VWF D′D3 assembly (Figure 1A), for which the IC was homozygous. Three family members, her father, mother, and younger sibling (II-1, II-2, III-3), were found to be heterozygous for the variant. Her older sibling (III-1) was found to be homozygous for the variant. The family pedigree is shown in Figure 1B. In silico analysis (Alamut Visual software; Interactive Biosoftware, version 2.9.0) suggests that this variant is damaging to protein expression and/or function (Figure 1C). Although this variant has not been documented by the European Association for Haemophilia and Allied Disorders VWF variant database, it was identified in 2 heterozygous males in the ThromboGenomics patient cohort.23 The variant has since been reported to the ClinVar database (https://www.ncbi.nlm.nih.gov/clinvar/; accessed 22 March 2021) as a “Variant of Uncertain Significance” with a minor allele frequency of 0.00082%. Two common benign VWF sequence variants were also identified in certain family members (supplemental Tables 1 and 2). No F8 sequence variants were identified in any family members.
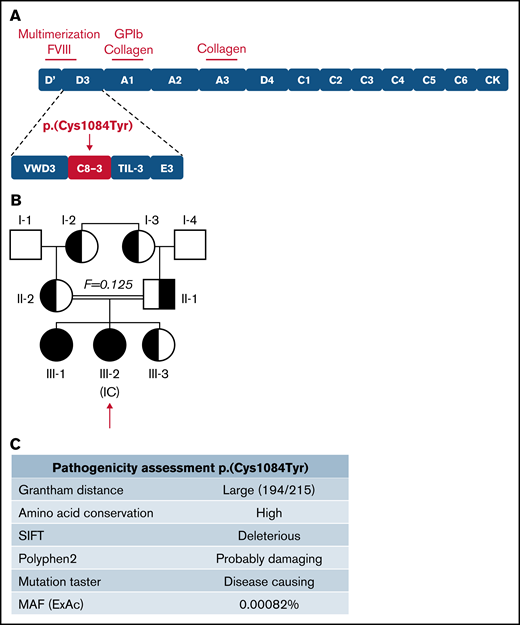
Inheritance and pathogenicity of novel VWF variant p.(Cys1084Tyr). (A) Schematic representation of the VWF domain structure with subdomain architecture and critical functional sites. The location of the p.(Cys1084Tyr) variant in the C8-3 subdomain is shown. (B) Three-generation pedigree of a consanguineous family diagnosed with VWD. Homozygous individuals are represented by black symbols and heterozygous individuals by half-filled symbols. (C) Pathogenicity prediction for VWF variant p.(Cys1084Tyr).
Inheritance and pathogenicity of novel VWF variant p.(Cys1084Tyr). (A) Schematic representation of the VWF domain structure with subdomain architecture and critical functional sites. The location of the p.(Cys1084Tyr) variant in the C8-3 subdomain is shown. (B) Three-generation pedigree of a consanguineous family diagnosed with VWD. Homozygous individuals are represented by black symbols and heterozygous individuals by half-filled symbols. (C) Pathogenicity prediction for VWF variant p.(Cys1084Tyr).
Phenotypic characterization of family members with the p.(Cys1084Tyr) variant
Family phenotypic values are outlined in Table 1. For 3 family members, heterozygosity for the p.(Cys1084Tyr) variant was associated with qualitative VWF defects. Normal VWF:Ag levels but significantly reduced VWF:GPIbM (<0.40 IU/mL) and VWF:CB (<0.35 IU/mL) were observed, consistent with a functional VWF defect and a diagnosis of type 2 VWD. Importantly, the clinical severity of the disease was markedly different among the heterozygous family members. Although the variant exhibited variable expressivity in the father (II-1) and the youngest sibling (III-3) who had normal bleeding scores, the mother (II-2) had a positive bleeding score (BS 8). The well-established quantitative effect of ABO blood group on plasma VWF levels likely contributes to the more elevated VWF:Ag levels observed in the father (II-1, blood group A) but does not account for absence of a bleeding diathesis given the qualitative nature of the defect.24,25 The prepubescent age (9 years) of the youngest sibling (III-3) may contribute at least in part to her normal bleeding score.
Laboratory and clinical phenotypic data for family members heterozygous and homozygous for p.(Cys1084Tyr)
| II-1 | II-2 | III-1 | III-2 (IC) | III-3 | Normal range | |
|---|---|---|---|---|---|---|
| Sex | M | F | F | F | F | − |
| Age | 35 | 41 | 10 | 9 | 3 | − |
| Genotype | c.[3251G>A];[=] | c.[3251G>A];[=] | c.[3251G>A];[3251G>A] | c.[3251G>A];[3251G>A] | c.[3251G>A];[=] | − |
| BAT score | 0 | 8 | 4 | 4 | 0 | 0-4 (adult), 0-2 (pediatric) |
| VWF:Ag (IU/mL) | 0.94 | 0.63 | 0.38 | 0.23 | 0.64 | 0.50-1.50 (IU/mL) |
| VWF:GPIbM (IU/mL) | 0.41 | 0.32 | 0.15 | 0.11 | 0.29 | 0.50-1.50 (IU/mL) |
| VWF:CB (IU/mL) | 0.34 | 0.26 | 0.13 | 0.09 | 0.3 | 0.50-1.50 (IU/mL) |
| VWFpp (IU/mL) | 1.13 | 0.84 | 0.76 | 0.56 | 0.91 | 0.50-1.50 (IU/mL) |
| FVIII:C (IU/mL) | 0.65 | 0.83 | 0.10 | 0.05 | 0.72 | 0.50-1.50 (IU/mL) |
| VWF:GPIbM/VWF:Ag | 0.45 | 0.50 | 0.40 | 0.48 | 0.45 | 0.75-1.25 |
| VWF:CBA/VWF:Ag | 0.38 | 0.41 | 0.34 | 0.40 | 0.47 | 0.75-1.25 |
| VWFpp/VWF:Ag | 1.26 | 1.33 | 2.01 | 2.47 | 1.42 | −2.2 |
| FVIII:C/VWF:Ag | 0.72 | 1.31 | 0.25 | 0.22 | 1.12 | 0.75-1.25 |
| Blood Group | A | O | O | O | O | − |
| II-1 | II-2 | III-1 | III-2 (IC) | III-3 | Normal range | |
|---|---|---|---|---|---|---|
| Sex | M | F | F | F | F | − |
| Age | 35 | 41 | 10 | 9 | 3 | − |
| Genotype | c.[3251G>A];[=] | c.[3251G>A];[=] | c.[3251G>A];[3251G>A] | c.[3251G>A];[3251G>A] | c.[3251G>A];[=] | − |
| BAT score | 0 | 8 | 4 | 4 | 0 | 0-4 (adult), 0-2 (pediatric) |
| VWF:Ag (IU/mL) | 0.94 | 0.63 | 0.38 | 0.23 | 0.64 | 0.50-1.50 (IU/mL) |
| VWF:GPIbM (IU/mL) | 0.41 | 0.32 | 0.15 | 0.11 | 0.29 | 0.50-1.50 (IU/mL) |
| VWF:CB (IU/mL) | 0.34 | 0.26 | 0.13 | 0.09 | 0.3 | 0.50-1.50 (IU/mL) |
| VWFpp (IU/mL) | 1.13 | 0.84 | 0.76 | 0.56 | 0.91 | 0.50-1.50 (IU/mL) |
| FVIII:C (IU/mL) | 0.65 | 0.83 | 0.10 | 0.05 | 0.72 | 0.50-1.50 (IU/mL) |
| VWF:GPIbM/VWF:Ag | 0.45 | 0.50 | 0.40 | 0.48 | 0.45 | 0.75-1.25 |
| VWF:CBA/VWF:Ag | 0.38 | 0.41 | 0.34 | 0.40 | 0.47 | 0.75-1.25 |
| VWFpp/VWF:Ag | 1.26 | 1.33 | 2.01 | 2.47 | 1.42 | −2.2 |
| FVIII:C/VWF:Ag | 0.72 | 1.31 | 0.25 | 0.22 | 1.12 | 0.75-1.25 |
| Blood Group | A | O | O | O | O | − |
In addition to the qualitative defects, homozygosity for the p.(Cys1084Tyr) variant was further associated with severely reduced plasma FVIII:C (<0.10 IU/mL) and a quantitative VWF defect. Plasma VWF:Ag levels were significantly reduced (<0.40 IU/mL) and FVIII:C/VWF:Ag in line with a diagnosis of type 2N were observed. Additionally, both homozygous siblings had positive bleeding scores. Taken together, the phenotypic data reveal that the p.(Cys1084Tyr) variant causes variable expressivity of a qualitative form of VWD in the heterozygous family members but results in a fully expressed VWD phenotype in the homozygous individuals who exhibit a more complex condition involving both qualitative and quantitative defects.
Reduction of HMWMs in individuals homozygous for p.(Cys1084Tyr)
To distinguish whether the qualitative aspect of the clinical phenotype was consistent with type 2A or type 2M VWD, VWF multimer analysis was performed on patient plasma (Figure 2A). Densitometric analysis revealed a relative reduction in HMWMs compared with normal pooled plasma for both the heterozygous and homozygous family members (Figure 2B). The reduction in HMWMs was most pronounced in the IC (III-2). This aberrant multimer pattern is characteristic of type 2A VWD and reflects the reduced collagen-binding activity observed in both heterozygous and homozygous family members (Table 1).
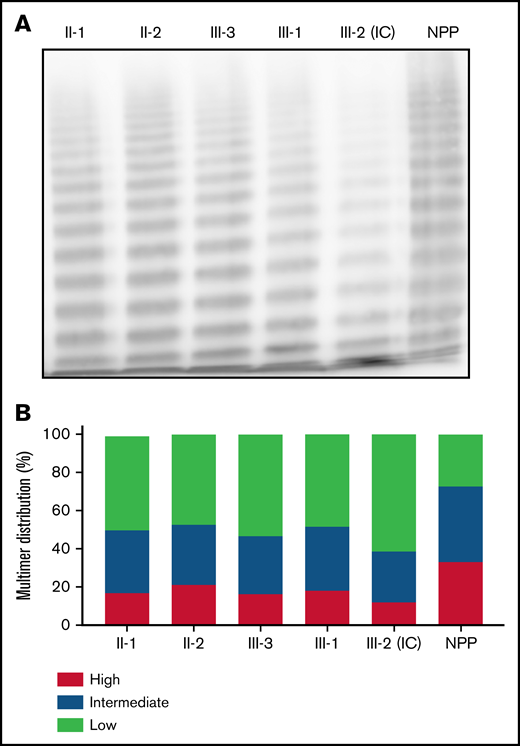
Phenotypic characterization of family members. (A) Multimer distribution of plasma VWF in heterozygous and homozygous family members relative to normal pooled plasma (NPP). Heterozygous and, to a greater degree, homozygous family members show a relative reduction in high molecular weight multimers. (B) Multimer analysis was performed using 1-dimensional densitometry and the relative percentage of high-, intermediate-, and low-molecular-weight multimers was reported. The greatest reduction in high-molecular-weight material was observed in the IC.
Phenotypic characterization of family members. (A) Multimer distribution of plasma VWF in heterozygous and homozygous family members relative to normal pooled plasma (NPP). Heterozygous and, to a greater degree, homozygous family members show a relative reduction in high molecular weight multimers. (B) Multimer analysis was performed using 1-dimensional densitometry and the relative percentage of high-, intermediate-, and low-molecular-weight multimers was reported. The greatest reduction in high-molecular-weight material was observed in the IC.
p.(Cys1084Tyr) VWF is rapidly cleared after desmopressin administration
To investigate the pathogenic mechanisms associated with p.(Cys1084Tyr), VWF:Ag, VWF:GPIbM, and FVIII:C plasma levels were measured in the homozygous siblings after desmopressin administration (Table 2). Although peak postdesmopressin VWF:Ag levels were elevated approximately threefold over baseline and normalized (>0.5 IU/mL) after 1 hour in both cases, the observed increases in plasma VWF:Ag levels were not sustained (Figure 3A-B). VWF:Ag half-lives were significantly reduced compared with the 12- to 18-hour half-life described in healthy individuals and were calculated as 4.20 and 6.41 hours for the IC (III-2) and her sibling (III-1), respectively.26 Consistently, a marked reduction in postdesmopressin VWF:GPIbM survival was also observed (IC III-2, t1/2 2.40 hours; III-3, t1/2 4.27 hours).
Pre- and postdesmopressin clinical laboratory values for homozygous family members
| Laboratory values | III-1 | III-2 (IC) |
|---|---|---|
| Base-line VWF:Ag (IU/mL) | 0.26 | 0.20 |
| Peak VWF:Ag (IU/mL) | 0.79 | 0.67 |
| VWF:Ag half-life (hours) | 6.41 | 4.20 |
| Base-line VWF:GP1bM (IU/mL) | 0.16 | 0.10 |
| Peak VWF:GP1bM (IU/mL) | 0.72 | 0.64 |
| VWF:GP1bM half-life (hours) | 4.27 | 2.40 |
| Base-line FVIII:C (IU/mL) | 0.05 | 0.04 |
| Peak FVIII:C (IU/mL) | 0.62 | 0.61 |
| FVIII:C half-life (IU/mL) | 1.90 | 1.05 |
| Laboratory values | III-1 | III-2 (IC) |
|---|---|---|
| Base-line VWF:Ag (IU/mL) | 0.26 | 0.20 |
| Peak VWF:Ag (IU/mL) | 0.79 | 0.67 |
| VWF:Ag half-life (hours) | 6.41 | 4.20 |
| Base-line VWF:GP1bM (IU/mL) | 0.16 | 0.10 |
| Peak VWF:GP1bM (IU/mL) | 0.72 | 0.64 |
| VWF:GP1bM half-life (hours) | 4.27 | 2.40 |
| Base-line FVIII:C (IU/mL) | 0.05 | 0.04 |
| Peak FVIII:C (IU/mL) | 0.62 | 0.61 |
| FVIII:C half-life (IU/mL) | 1.90 | 1.05 |
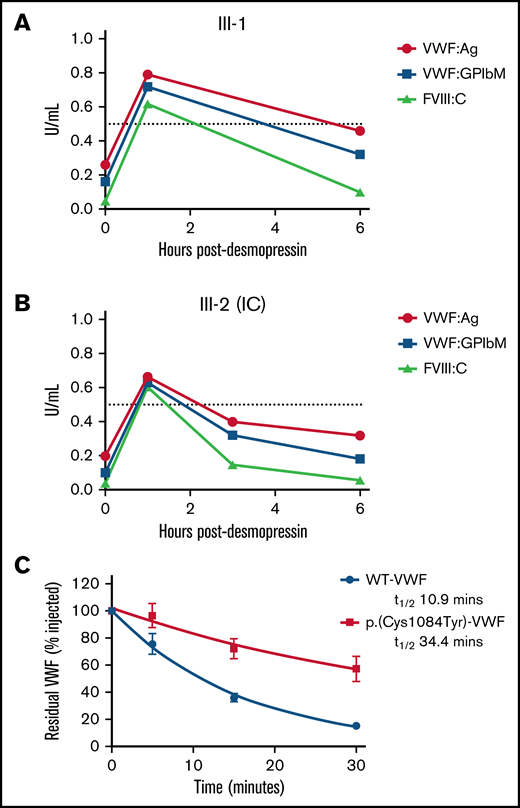
Clearance kinetics of p.(Cys1084Tyr)-VWF. Time course of the postdesmopressin response in siblings III-1 (A) and III-2 (B). Markedly reduced VWF:Ag, VWF:GPIbM and FVIII:C half-lives were observed following the 1-hour peak. The dashed line represents normalized VWF:Ag, VWF:GPIbM and FVIII:C plasma levels (0.5 IU/mL). (C) The influence of p.(Cys1084Tyr) on VWF clearance in a mouse model of VWD. Prolonged survival of p.(Cys1084Tyr)-VWF was observed. Data were fitted to a 1-phase exponential decay (n = 3 mice per time point).
Clearance kinetics of p.(Cys1084Tyr)-VWF. Time course of the postdesmopressin response in siblings III-1 (A) and III-2 (B). Markedly reduced VWF:Ag, VWF:GPIbM and FVIII:C half-lives were observed following the 1-hour peak. The dashed line represents normalized VWF:Ag, VWF:GPIbM and FVIII:C plasma levels (0.5 IU/mL). (C) The influence of p.(Cys1084Tyr) on VWF clearance in a mouse model of VWD. Prolonged survival of p.(Cys1084Tyr)-VWF was observed. Data were fitted to a 1-phase exponential decay (n = 3 mice per time point).
Additionally, the relative FVIII:C response was significant (15- and 12-fold increase over baseline for III-2 (IC) and III-1, respectively), and the absolute peak postdesmopressin FVIII:C values were in the normal range. In keeping with the reduced VWF:Ag survival, significantly shortened FVIII:C survival was also apparent (III-2 [IC], t1/2 1.05 hours; III-3, t1/2 1.90 hours). Moreover, the shorter half-life of FVIII:C compared with VWF:Ag confirmed the presence of a FVIII-binding defect in these individuals. Collectively, the desmopressin response kinetics demonstrate that both accelerated VWF clearance and defective FVIII binding contribute to VWD pathophysiology in family members homozygous for the p.(Cys1084Tyr) variant.
Prolonged survival of p.(Cys1084Tyr)-VWF in VWF−/− mice
To investigate the mechanisms underlying rapid clearance of p.(Cys1084Tyr)-VWF, recombinant VWF infusions were carried out in VWF−/− mice. Surprisingly, the half-life of p.(Cys1084Tyr)-VWF was significantly prolonged compared with WT-VWF (t1/2 34.4 vs 10.9 minutes, respectively; P < .0001, Figure 3C). Although human and mouse VWF share ∼80% amino acid identity and several cysteine VWF variants have previously been shown to undergo rapid clearance in this mouse model, it is apparent that the rapid clearance of p.Cys1084Tyr-VWF was not recapitulated in this setting.13,27 The significance of this finding and the underlying molecular mechanisms remain unclear.
p.(Cys1084Tyr) is associated with reduced VWF secretion and aberrant multimerization in vitro
To assess the impact of the p.(Cys1084Tyr) variant on VWF synthesis and secretion, HEK 293T cells were transiently transfected with full-length human VWF expression vectors encoding WT or the p.(Cys1084Tyr) variant to mimic the heterozygous and homozygous states. For cells transfected with the cDNA coding for the p.(Cys1084Tyr) variant alone, significantly reduced VWF secretion was observed in the conditioned media (25.2 ± 7.0% of WT-VWF; P < .0001; Figure 4A-B). For cotransfected cells, a less severe reduction in VWF secretion was observed (58.4 ± 5.2% of WT-VWF; P < .0001), indicating that the p.(Cys1084Tyr) variant negatively affected VWF secretion in a dose-dependent manner.
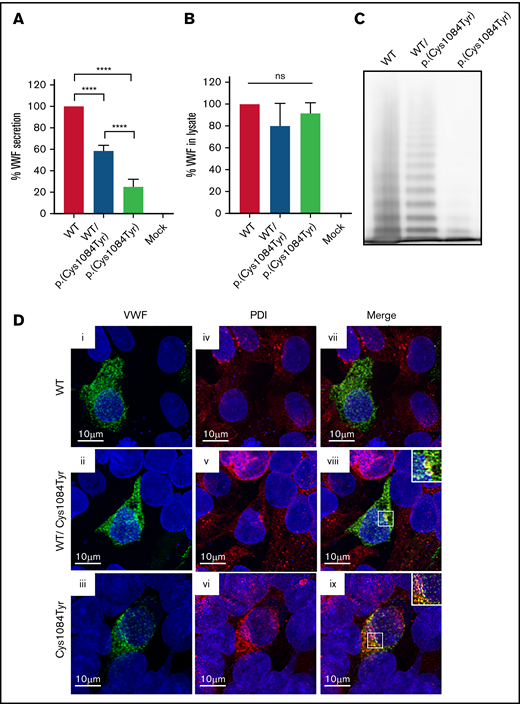
Defective secretion despite normal storage of p.(Cys1084Tyr) VWF in vitro. (A) HEK293T cells were transiently transfected with cDNA encoding WT-VWF, the p.(Cys1084Tyr) variant, or a 1:1 molar ratio of both constructs to mimic the heterozygous state. Transfection with cDNA encoding the p.(Cys1084Tyr) variant led to a significant reduction in the amount of VWF secreted into the conditioned media whether transfected alone or cotransfected with the cDNA coding for WT-VWF. The reduction in VWF secretion was more severe for the transfection mimicking the homozygous condition. Data are represented as mean ± standard error of the mean (SEM), ****P < .0001. (B) No increase in variant VWF was observed in the cellular lysate for either the heterozygous or homozygous state indicating that the p.(Cys1084Tyr) variant does not cause intracellular retention of VWF. (C) Multimer analysis of recombinant VWF showed a relative reduction in high-molecular-weight multimers for VWF produced via cotransfection and a complete absence of high- and intermediate-weight multimers for p.(Cys1084Tyr) VWF. (D) Pseudo-WPB formation was assessed via transient transfection in HEK293 cells. Transfected cells were fixed and stained for VWF (green; i, ii, iii) and resident ER protein protein disulfide isomerase (PDI) (red; iv, v, vi). Merged images (vii, viii, ix) are shown in the final column. For all images, blue indicates 4′,6-diamidino-2-phenylindole. Colocalization of VWF and PDI was observed for cotransfected cells (viii) and for cells transfected with p.(Cys1084Tyr)-VWF alone (ix). Magnified views of regions of colocalization are shown in the inset. Images are representative of n = 3 independent experiments.
Defective secretion despite normal storage of p.(Cys1084Tyr) VWF in vitro. (A) HEK293T cells were transiently transfected with cDNA encoding WT-VWF, the p.(Cys1084Tyr) variant, or a 1:1 molar ratio of both constructs to mimic the heterozygous state. Transfection with cDNA encoding the p.(Cys1084Tyr) variant led to a significant reduction in the amount of VWF secreted into the conditioned media whether transfected alone or cotransfected with the cDNA coding for WT-VWF. The reduction in VWF secretion was more severe for the transfection mimicking the homozygous condition. Data are represented as mean ± standard error of the mean (SEM), ****P < .0001. (B) No increase in variant VWF was observed in the cellular lysate for either the heterozygous or homozygous state indicating that the p.(Cys1084Tyr) variant does not cause intracellular retention of VWF. (C) Multimer analysis of recombinant VWF showed a relative reduction in high-molecular-weight multimers for VWF produced via cotransfection and a complete absence of high- and intermediate-weight multimers for p.(Cys1084Tyr) VWF. (D) Pseudo-WPB formation was assessed via transient transfection in HEK293 cells. Transfected cells were fixed and stained for VWF (green; i, ii, iii) and resident ER protein protein disulfide isomerase (PDI) (red; iv, v, vi). Merged images (vii, viii, ix) are shown in the final column. For all images, blue indicates 4′,6-diamidino-2-phenylindole. Colocalization of VWF and PDI was observed for cotransfected cells (viii) and for cells transfected with p.(Cys1084Tyr)-VWF alone (ix). Magnified views of regions of colocalization are shown in the inset. Images are representative of n = 3 independent experiments.
Multimer analysis of the secreted VWF demonstrated aberrant multimerization for the homozygous state that was significantly more severe than that observed in patient plasma. The complete absence of both HMWMs and intermediate-molecular-weight multimers was observed, with only the lowest-molecular-weight VWF multimers visible (Figure 4C). This phenomenon has previously been reported and likely represents cell-specific differences in the posttranslational and quality control pathways in endothelial vs nonendothelial cells.28
To visualize pseudo-WPB formation, transfections were repeated using HEK 293 cells. Imaging studies revealed that the p.(Cys1084Tyr) variant affects packaging and storage of VWF into pseudo-WPBs (Figure 4D). Cells transfected with WT-VWF formed distinct, cigar-shaped pseudo-WPBs, as did cotransfected cells, whereas cells transfected with the p.(Cys1084Tyr) variant alone displayed both granular and diffuse staining. Furthermore, p.(Cys1084Tyr)-VWF staining overlapped with that of ER marker PDI, suggesting that at least a portion of the mutant subunits were retained within the ER. Colocalization was also observed for cotransfected cells, although to a lesser extent. Taken together, these data demonstrate that packaging and secretion of p.(Cys1084Tyr)-VWF is impaired in both the homozygous and heterozygous states.
p.(Cys1084Tyr) abrogates VWF secretion in a mouse model of VWD
To characterize the in vivo effects of the p.(Cys1084Tyr) substitution, we sought to develop a mouse model of this complex form of VWD. Accordingly, in vivo VWF expression was induced via hydrodynamic tail vein injection of the murine Vwf cDNA with or without the variant coding for p.(Cys1084Tyr) in VWF−/− mice.
In keeping with previous reports, variable supraphysiologic VWF expression was observed in the plasma of VWF−/− mice that received WT mouse Vwf cDNA (VWF:Ag, 17.5 ± 6.4U/mL).29 In contrast, no VWF was detectable in the plasma of VWF−/− mice that received mouse Vwf cDNA coding for the p.(Cys1084Tyr) variant (Figure 5A). Plasma VWF propeptide was also undetectable in these mice (data not shown). However, immunohistochemical DAB staining of mouse liver sections revealed robust VWF expression in a population of hepatocytes. Intracellular hepatocyte p.(Cys1084Tyr)-VWF expression was comparable to that observed for WT-VWF, with approximately 5% of hepatocytes expressing WT or variant VWF (Figure 5B). In keeping with our in vitro imaging results, in these transient transgenic in vivo studies, p.(Cys1084Tyr)-VWF was synthesized by mouse hepatocytes but was not efficiently secreted into plasma, suggesting compromised VWF secretion. The absence of detectable plasma VWF in this mouse model of VWD precluded any further investigation of the bleeding phenotype associated with this cysteine variant.
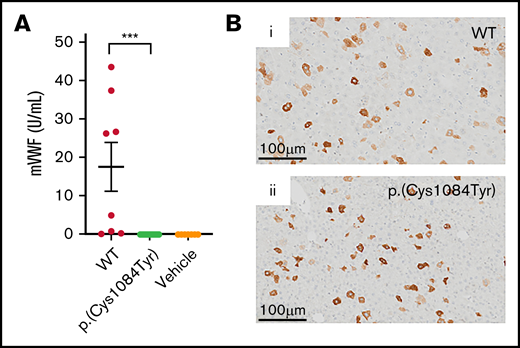
Hepatic expression of p.(Cys1084Tyr) abolishes VWF secretion in VWF−/− mice. (A) Following hydrodynamic gene transfer of the cysteine variant mouse Vwf cDNA, no p.(Cys1084Tyr) VWF was detectable in the plasma of VWF−/− mice. Data represent mean ± SEM, ***P < .001. (B) DAB peroxidase staining of formalin-fixed paraffin-embedded mouse liver sections showed equivalent intracellular expression of both WT-VWF and p.(Cys1084Tyr) VWF in ∼5% of hepatocytes.
Hepatic expression of p.(Cys1084Tyr) abolishes VWF secretion in VWF−/− mice. (A) Following hydrodynamic gene transfer of the cysteine variant mouse Vwf cDNA, no p.(Cys1084Tyr) VWF was detectable in the plasma of VWF−/− mice. Data represent mean ± SEM, ***P < .001. (B) DAB peroxidase staining of formalin-fixed paraffin-embedded mouse liver sections showed equivalent intracellular expression of both WT-VWF and p.(Cys1084Tyr) VWF in ∼5% of hepatocytes.
Reduced VWF secretion is caused by loss of the p.(Cys1060)-p.(Cys1084) disulfide bond
Evidence exists that the loss of a disulfide bond because of mutation of a cysteine residue and the subsequent effect on protein folding is of greater consequence to VWF expression than the generation of the associated novel free thiol.15 In WT-VWF an intra-subunit disulfide bond connects p.(Cys1084) to p.(Cys1060) in the C8-3 subdomain, linking and stabilizing the α1 and α3 helices (Figure 6A-B).5 Consequently, in patients with the p.(Cys1084Tyr) variant, p.(Cys1060) is predicted to exist as a free thiol (Figure 6A). To assess the impact of the novel free thiol on VWF secretion, serine (conservative) or alanine (nonconservative) p.(Cys1060;Cys1084) double variants were generated via site-directed mutagenesis. Individual removal of p.(Cys1084) or p.(Cys1060) lead to significant reductions in the amount of VWF detected in conditioned media: p.(Cys1084Tyr), 25.2 ± 7.0%; p.(Cys1060Ala), 37.4 ± 13.0%; p.(Cys1060Ser), 11.5 ± 2.1% (WT-VWF; P < .01; Figure 7A-B). However, elimination of the novel free thiol via simultaneous removal of both p.(Cys1060) and p.(Cys1084) did not rescue VWF secretion: p.(Cys1060A;p.Cys1084Tyr, 19.5 ± 1.9% and p.(Cys1060S;p.Cys1084Tyr), 15.9 ± 2.4% (WT- VWF; P < .0001).
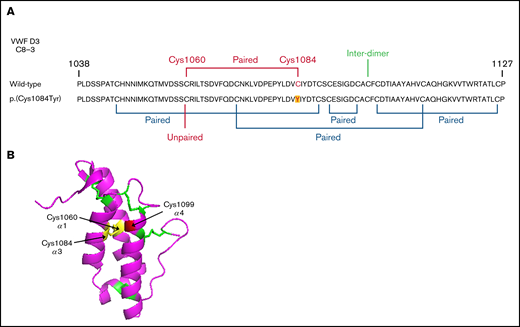
Disulfide connectivity and structural architecture of the VWF C8-3 subunit. (A) Disulfide connectivity of the C8-3 subdomain of the VWF D′D3 assembly. (B) Location of p.(Cys1084) in the crystal structure of the VWF C8-3 subdomain. Intra-subdomain disulfide bonds are annotated in green, the p.(Cys1060)-p.(Cys1084) disulfide bond in yellow, and the cysteine involved in inter-subunit bonding p.(Cys1099) in red. Protein Data Bank 6N29, accessed 20 March 2021. Image rendering was performed with PyMOL software (version 2.1.1).
Disulfide connectivity and structural architecture of the VWF C8-3 subunit. (A) Disulfide connectivity of the C8-3 subdomain of the VWF D′D3 assembly. (B) Location of p.(Cys1084) in the crystal structure of the VWF C8-3 subdomain. Intra-subdomain disulfide bonds are annotated in green, the p.(Cys1060)-p.(Cys1084) disulfide bond in yellow, and the cysteine involved in inter-subunit bonding p.(Cys1099) in red. Protein Data Bank 6N29, accessed 20 March 2021. Image rendering was performed with PyMOL software (version 2.1.1).
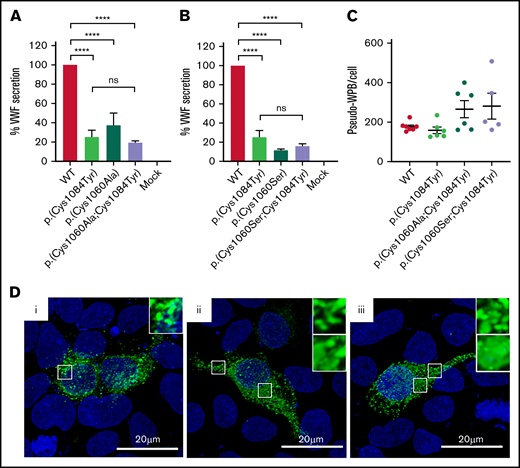
Altered disulfide connectivity because of loss of the p.(Cys1060)-p.(Cys1084) disulfide bond causes reduced VWF secretion. (A) Transient transfection of HEK293 cells with the expression vector encoding p.(Cys1060Ala) caused a significant reduction in VWF secretion. Transfection of cells with the expression vector coding for the p.(Cys1060Ala;Cys1084Tyr) double variant did not rescue VWF secretion. (B) Similar results were observed for p.(Cys1060Ser) and the p.(Cys1060Ser;Cys1084Tyr) double variant. Data are represented as mean ± SEM, ****P < .0001. (C) Three-dimensional quantification of the number of pseudo-WPB/cell revealed no significant difference in the number of storage organelles formed/cell by each variant. (D) Immunofluorescent imaging of WT-VWF (i), p.(Cys1060Ala;Cys1084Tyr) (ii), and p.(Cys1060Ser;Cys1084Tyr) (iii). VWF is depicted in green, and blue represents 4′,6-diamidino-2-phenylindole. Elongated rod-shaped WPBs are shown (inset, top). Areas of diffuse VWF staining for p.(Cys1060Ala;Cys1084Tyr) and p.(Cys1060Ser;Cys1084Tyr) indicating impaired regulated storage are also shown (inset, bottom). Images are representative of n = 3 independent experiments.
Altered disulfide connectivity because of loss of the p.(Cys1060)-p.(Cys1084) disulfide bond causes reduced VWF secretion. (A) Transient transfection of HEK293 cells with the expression vector encoding p.(Cys1060Ala) caused a significant reduction in VWF secretion. Transfection of cells with the expression vector coding for the p.(Cys1060Ala;Cys1084Tyr) double variant did not rescue VWF secretion. (B) Similar results were observed for p.(Cys1060Ser) and the p.(Cys1060Ser;Cys1084Tyr) double variant. Data are represented as mean ± SEM, ****P < .0001. (C) Three-dimensional quantification of the number of pseudo-WPB/cell revealed no significant difference in the number of storage organelles formed/cell by each variant. (D) Immunofluorescent imaging of WT-VWF (i), p.(Cys1060Ala;Cys1084Tyr) (ii), and p.(Cys1060Ser;Cys1084Tyr) (iii). VWF is depicted in green, and blue represents 4′,6-diamidino-2-phenylindole. Elongated rod-shaped WPBs are shown (inset, top). Areas of diffuse VWF staining for p.(Cys1060Ala;Cys1084Tyr) and p.(Cys1060Ser;Cys1084Tyr) indicating impaired regulated storage are also shown (inset, bottom). Images are representative of n = 3 independent experiments.
When transfections were repeated in HEK 293 cells to visualize pseudo-WPB formation, a combination of diffuse and granular VWF staining was observed for the double mutants, indicating impaired VWF packaging (Figure 7D). Considerable variation was also observed in the number of pseudo-WPBs formed per cell by each of the double mutants, although the overall number of pseudo-WPBs formed per cell did not significantly differ to WT-VWF (Figure 7C). Collectively, these data demonstrate that the effects of cysteine mutation on VWF secretion are primarily attributable to errant disulfide connectivity caused by the loss of the disulfide bond and subsequent protein misfolding rather than the presence of a free thiol.
Discussion
In this study, we identified and characterized a novel VWD-causing cysteine variant in the VWF D′D3 assembly: p.(Cys1084Tyr). This cysteine variant was found to cause an unusual, variably expressed, and phenotypically complex form of VWD involving intersecting type 1, type 2A, and type 2N phenotypes. Although VWF cysteine variants are known to contribute to the pathogenesis of both type 1 and type 2 VWD, instances of such phenotypic complexity and variability in a single kindred are rare. Extensive characterization of affected family members revealed that, although both heterozygosity and homozygosity for the p.(Cys1084Tyr) variant were associated with clinical bleeding, they were in fact associated with remarkably dissimilar diagnoses. Based on the recent American Society of Hematology/International Society on Thrombosis and Hemostasis/National Hemopilia Foundation/World Federation of Hemophilia VWD diagnostic criteria, family members who were heterozygous for the cysteine variant were diagnosed with type 2A VWD, whereas homozygous family members displayed a more severe and complex type 1/type 2A/type 2N phenotype.30 Unusually, clinical phenotypic variability was evident not only between homozygous and heterozygous family members but also among the heterozygous (type 2A) family members. Although the mechanistic complexity of type 2A VWD has been documented, with a number of variably influential defects collectively resulting in the overall disease phenotype, type 2A VWD is typically associated with largely consistent expressivity of specific VWF variants.31 In this family, however, the mother, who experienced the most severe bleeding symptoms (BS 8), exhibited a compromised VWF:GPIbM/VWF:Ag and a relative reduction in HMWMs, whereas the father, who had a comparable reduction in VWF:GPIbM/VWF:Ag and an equivalent multimerization defect, had a normal bleeding score. The reason for this disparity is unclear. However, previous studies have attributed phenotypic variability in type 2 VWD to the variable multimer composition of circulating plasma VWF.32 Consequently, the extent of p.(Cys1084Tyr) monomer incorporation into the final circulating multimer of plasma VWF may contribute to the variable expressivity observed in the heterozygous family members.
The detrimental effect of substituting cysteine residues involved in VWF dimerization or multimerization is intuitive. However, the outcomes associated with substitution of other VWF cysteine residues are less predictable, as specific roles for most disulfides in maintaining VWF structure/function have not been definitively determined. The recent resolution of the VWF D′D3 crystal structure has enabled better prediction of these outcomes and provides valuable insight into some of the molecular mechanisms by which variants such as the p.(Cys1084Tyr) substitution cause VWD.5
The location of the p.(Cys1060)-p.(Cys1084) disulfide bond within 1 of the 2 noncontiguous FVIII binding sites, as well as its close proximity to p.(Cys1099), a critical residue for VWF multimerization, reveals the mechanistic basis by which p.(Cys1084Tyr) can impact both FVIII binding and VWF multimerization (and thus GpIb and collagen binding).5,33 However, the mechanisms governing accelerated clearance of p.(Cys1084Tyr)-VWF are not readily apparent from examination of the crystal structure. Although pathologic clearance has previously been reported for several cysteine variants in the VWF D3 domain, a detailed understanding of the underlying process has not been resolved.13 However, it seems plausible that rapid clearance of these variants involves a common mechanism of conformational distortion caused by the loss of key structural cysteine residues with subsequent exposure of otherwise cryptic clearance receptor-interactive sites. Interestingly, unlike other D3 domain cysteine variants, the standard mouse model of VWF clearance has not proven informative in the context of the p.Cys1084Tyr variant.13 However, given the high degree of amino acid identity between the human and murine VWF D′D3 assemblies (∼89%) and between the smaller C8-3 subdomains (∼91%), it is likely that differences between murine and human forms of key VWF clearance receptors are responsible for the discordant p.Cys1084Tyr clearance profiles. To date, several limitations of the standard mouse model of VWF clearance have been acknowledged. For example, in humans, differential VWF clearance based on ABO(H) blood group status is well documented.25,34,35 However, following infusion of blood group–specific human VWF, this differential ABO(H) effect is not observed in mice.36 In addition, murine scavenger receptor stabilin-2 has been shown to function as a clearance receptor for human but not murine VWF.22 Conversely, murine stabilin-2 clears murine but not human VWF propeptide.37 The mechanistic basis for this is not currently understood but emphasizes the complex, multifactorial nature of VWF clearance and the clear differences between VWF clearance pathways in humans and mice.
Another aspect of the multifaceted phenotype that cannot be further elucidated by examination of the D′D3 crystal structure is the impact of the p.(Cys1084Tyr) variant on VWF secretion. We demonstrated impaired secretion of the p.(Cys1084Tyr) variant both in vitro and in vivo using a heterologous expression system and a transient transgenic mouse model. Notwithstanding this observation, the magnitude of the postdesmopressin response in homozygous individuals provides evidence for the presence of a releasable VWF pool. Taken together, these data suggest that the p.(Cys1084Tyr) variant does not affect stimulated VWF secretion; rather, p.(Cys1084Tyr)-VWF has adverse effects on unstimulated VWF secretion. Unstimulated VWF secretion refers to 2 different secretory pathways: the basal and constitutive VWF secretory pathways. Basal secretion refers to the spontaneous release of VWF from regulated storage in WPBs in the absence of stimulation, whereas constitutive secretion refers to the continuous transfer of nascent VWF from the Golgi network to the outside surface of the cell. Because the current consensus advises that the majority (∼80%) of plasma VWF arises from basal secretion from WPBs, and the p.(Cys1084Tyr) variants impairs packing of VWF into these specialized organelles, the question arises if basal but not regulated release of this particular cysteine variant could be compromised, leading to the reduced VWF plasma levels observed in affected individuals.38
We further extended our observations regarding p.(Cys1084Tyr)-VWF secretion to demonstrate that impaired secretion resulted from the loss of the disulfide bond rather than the presence of the novel free thiol. Importantly, however, this does not negate the possibility that the presence of the free thiol may contribute to other aspects of the complex phenotype. Variants at both p.(Cys1060) and p.(Cys1084) have been reported to cause VWD.11 Although homozygosity for p.(Cys1084Tyr) is associated with the complex mixed phenotype described herein, previous reports have described type 2N VWD in a patient homozygous for p.(Cys1060Arg).39 Homozygous loss of p.(Cys1060) was associated with a dramatic reduction in FVIII-binding ability and a relative loss of HMWMs. However, VWF:Ag and VWF:RCo were normal. In light of the observation that both cases of VWD are associated with the disruption of the same intra-subunit disulfide bond, it is possible that the phenotypic differences observed for p.(Cys1060Arg) and p.(Cys1084Tyr) may reflect the relative proximity of the novel free thiol to, and potential interference with, critical functional sites (Figure 6A-B). Differential disulfide rearrangements arising from p.(Cys1084Tyr) vs p.(Cys1060Arg) may also have more profound effects on VWF structure and function.
The data herein serve to highlight the previously underappreciated complexities of cysteine variant VWD. Importantly, our study also provides insight into the limitations of the VWD classification system. Under the current VWD classification criteria, the heterozygous and homozygous members of this family are differentially categorized. The heterozygous family members are easily classified; however, the phenotypic classification of the homozygous individuals proves more difficult. At present, no definitive guidance exists on the appropriate classification of mixed phenotype forms of VWD. Thus, the questions arise as to whether this should be based on the most dominant phenotype, whether this can this be unambiguously established in every case, and whether effective clinical management requires absolute classification of the disease. As our understanding of VWF biology continues to evolve, so too will the complexity of classifying the condition based on discrete phenotypes.
Acknowledgments
The authors thank G. Jones and S. Tinlin of the National Inherited Bleeding Disorder Genotyping Laboratory, L. Boudreau and S. Virk of Queen's Laboratory for Molecular Pathology, and P. Lima from Queen’s CardioPulmonary Unit for technical assistance.
The study was supported by a foundation grant from the Canadian Institutes of Health Research (FDN154285). D.L. is the recipient of a Canada Research Chair in Molecular Hemostasis.
Authorship
Contribution: O.R. and L.L.S. designed and performed experiments and interpreted results; C.B. and K.N. performed experiments; O.R. wrote the manuscript; L.L.S., D.L., and P.D.J. edited the manuscript; M.R. performed the expert-administered BAT; and R.K., M.R., T.H., and M.D.C. recruited study subjects and performed sample collection.
Conflict-of-interest disclosure: O.R. has received research funding and honoraria for participating on advisory boards for CSL Behring. P.D.J. receives research funding from Bayer, Takeda and CSL Behring. M.D.C. has received research support from Bayer, Bioverativ/Sanofi, CSL-Behring, Novo Nordisk, Octapharma, Pfizer, and Shire and has received honoraria for speaking/participating on advisory boards for Bayer, Bioverativ/Sanofi, Biotest, CSL Behring, Grifols, LFB, Novo Nordisk, Octapharma, Pfizer, Roche, and Shire. D.L. has received research funding from Shire, Bayer, Bioverativ, CSL Behring, and Octapharma. The remaining authors declare no competing financial interests.
Correspondence: David Lillicrap, Richardson Laboratory, Queen's University, 88 Stuart Street, Kingston, ON K7L 3N6; e-mail: david.lillicrap@queensu.ca.

