

Key Points
IBR 420 mg daily in combination with VEN 200 mg daily were identified as having the highest response and lowest toxicty in relapsed MCL.
Additional benefit was not seen at higher doses. Resistance was seen at all dosing combinations, suggesting a biologic resistance mechanism.

Abstract
Relapsed Mantle cell lymphoma (MCL) is often treated with Bruton’s tyrosine kinase inhibitors (BTKi); however, post-BTKi relapse can be challenging. Adding venetoclax (VEN) to ibrutinib (IBR) has shown synergy in preclinical MCL models. Prior MCL studies of the combination show promising efficacy but have conducted limited dose finding. We sought to identify the optimal dosing combination, based on efficacy and toxicity, utilizing a continual reassessment method of 6 combinations of IBR (280 mg, 420 mg, and 560 mg by mouth daily) and VEN (max dose of 200 mg and 400 mg by mouth daily). Eligible participants were not previously exposed to BTKi and not high risk for tumor lysis syndrome (TLS). VEN, initiated first at 100 mg, then at 20 mg by mouth daily after a TLS event, was started prior to adding IBR and ramped-up based on the dose level assigned. Combination treatment continued for six 28-day cycles. Thirty-five participants were enrolled and treated. One TLS event occurred with starting dose of 100 mg VEN; no TLS was seen with 20 mg. The optimal dosing combination was considered to be VEN 200 mg and IBR 420 mg with an overall response rate (ORR) of 93.8% (95% CI: 73.6% to 99.7%) and DLT incidence of 6.2% (95% CI: 0.3% to 26.4%). ORR for all arms was 82.3% (28/34; 95% CI: 65.5% to 93.2%) with a complete response (CR) rate of 42.4% (14/33; 95% CI: 25.5% to 60.8%). A participant was not allocated to IBR 560 mg and VEN 400 mg. ORR benefit was not seen with higher dosing combinations and toxicity was higher; a comparison made within the limitations of small cohorts. Resistance was seen in nearly all arms. This trial was registered at www.clinicaltrials.gov #NCT02419560.
Introduction
Mantle cell lymphoma (MCL) is a rare form of non-Hodgkin lymphoma (NHL) with an incidence of around 4000 to 5000 cases per year1-3 in the United States. There are multiple histologic and clinical phenotypes ranging from an indolent, non-nodal subtype4-7 to an aggressive blastoid variety.2 MCL is typically treated with immunochemotherapy followed by high-dose chemotherapy with autologous stem cell rescue in transplant-eligible patients.8-12 However, despite aggressive therapy, a cure is seldom attained, with progression-free survival (PFS) ranging from a median of 7 to 9 years, depending on the induction treatment approach.8-11
Ibrutinib (IBR), a covalent, oral, small-molecule Bruton tyrosine kinase inhibitor (BTKi), has changed the landscape of MCL patients who relapse after immunochemotherapy. Treatment at a dose of 560 mg daily showed an overall response rate (ORR) of 67% and a 2-year PFS of 31%.13,14 At this dose, there is nearly 100% BTK occupancy.15 However, not all patients with relapsed or refractory MCL benefit, with 20% to 33% of patients failing to respond to single-agent therapy,13,14,16 and complete response (CR) only occurs in about a quarter of those treated. Relapse after IBR failure has a poor outcome.17,18 Mechanisms of resistance to IBR in MCL remain elusive and appear to differ19 from the BTK and Phospholipase C-ɣ (PLCɣ) mutations observed in chronic lymphocytic leukemia (CLL) treatment failures.20,21 To study ways to improve response to IBR, our group and others identified a synergistic combination of IBR and venetoclax (VEN) in preclinical models that informed the present study.22,23
BCL-2 is an antiapoptotic protein that is dysregulated in many B-cell malignancies, including MCL.24 VEN is a small molecule, oral BCL-2 inhibitor with a high affinity to BCL-2 over other antiapoptotic proteins.25 Early studies with VEN showed rapid tumor cell killing complicated by tumor lysis syndrome (TLS), particularly in patients with CLL, necessitating a dose ramp-up to mitigate TLS.26 VEN has activity as a single agent in MCL with a response rate of 75% in a phase 1 study.27 However, despite early response, responses were not long-lasting27 . VEN had activity at maximum dose ranges from 400 mg to 800 mg by mouth daily.
To date, IBR and VEN have been combined in 2 published MCL studies. AIM used the single-agent phase 2 dose of each drug (IBR 560 mg by mouth daily and VEN 400 mg by mouth daily) in 24 patients with relapsed/refractory disease.28 OAsIs combined obinutuzumab with IBR plus VEN in a staged, phase 1/2 manner. IBR was fixed at 560 mg by mouth daily, and VEN was given at 400 mg, 600 mg, and 800 mg daily in a dose-escalation strategy.29 However, as patients treated at higher doses experienced more transfusion support and, as the preclinical rationale showed no improvement in efficacy at doses higher than 400 mg, 400 mg by mouth daily was used as the phase 2 dose. Importantly, hematologic toxicity was common in relapsed or refractory patients, primarily thrombocytopenia and neutropenia. Both studies treated patients with IBR for 4 weeks prior to starting the VEN dose ramp-up.
Given the preclinical rationale for the combination of IBR and VEN as well as concerns for toxicity, as both drugs are metabolized via CYP3A, we employed a dose-finding study in relapsed and refractory MCL to identify the optimal combination (ClinicalTrials.gov identifier NCT02419560). VEN was initiated first, and IBR was added during the VEN dose ramp-up; thus achieving the planned maximum dose earlier in the treatment course. We hypothesized that doses lower than the single-agent dose could achieve similar efficacy with limited toxicity. A continual reassessment method (CRM) was used to identify the optimal dose combination, defined as the combination of doses, which resulted in a dose-limiting toxicity (DLT) rate ≤25% and maximized the ORR (complete + partial remission) at 2 months posttreatment.
Methods
Patients
Eligible patients were ≥18 years of age, diagnosed with MCL (defined as the presence of cyclin D1 expression and/or t(11;14) by fluorescence in-situ hybridization [FISH]) and relapsed after 1 or more chemotherapy-containing regimens. Prior treatment with a BTKi was not allowed. Potential participants must have had radiographically measurable disease defined as at least 1 lymph node (LN) ≥2 cm in longest diameter or splenomegaly >13 cm in craniocaudal dimension. Participants at high risk for tumor lysis, defined as having active TLS, a measurable LN ≥10 cm, or a LN ≥5cm and an ALC (absolute lymphocyte count) >25 × 109/L, were excluded. Adequate organ function, including a calculated or measured creatinine clearance of ≥50 mL/min, was required. Potential participants who required treatment with CYP3A inhibitors or potent inducers or required warfarin for anticoagulation were excluded. Full eligibility criteria can be found in the supplemental Methods.
Treatment
A CRM was used to identify the optimal dose combination of IBR and VEN among 6 different dosing combinations. IBR was tested at 3 dosing levels (280 mg, 420 mg, and 560 mg by mouth daily), and VEN was tested at 2 dosing levels (max dose of 200 mg by mouth daily and max dose of 400 mg by mouth daily) (Table 1). The dosing schema is shown in Figure 1. Participants were initially treated with 100 mg of VEN daily for 1 week before starting IBR at the allocated dose. However, a clinically significant TLS event occurred (acute kidney injury with grade 4 hyperkalemia, hyperphosphatemia, and hyperuricemia occurring within 24 hours of the first dose of VEN 100 mg) in 1 of the first 15 patients treated, as previously reported.30 This event necessitated a pause in enrollment and a change to the initial dosing of venetoclax to mitigate TLS. After study amendment, patients were treated with VEN daily dosing of 20 mg for 2 days, 50 mg for 5 days, and 100 mg for 7 days (dosing windows were allowed) before starting the allocated dose of IBR. IBR plus 100 mg of VEN was continued for 1 week before further escalating VEN to the maximum allocated dose as per Figure 1. TLS was closely monitored during the VEN dose ramp-up. The combination of IBR and VEN continued for six 28-day cycles. Treatment after completing combination therapy was per treating physician discretion, though the protocol did suggest continuing single-agent IBR at 560 mg by mouth daily unless otherwise contraindicated. VEN was not provided by the study beyond cycle 6. Participants were followed for progression and survival.
Dosing allocation and outcomes
| All subjects 20-100 VEN (cycle 0) | IBR (week 1+) mg per day | |||
|---|---|---|---|---|
| 280 | 420 | 560 | ||
| VEN (mg per day) | 400 (week 3+) 200 (week 2) 100 (week 1) | Zone 2/arm C (n = 8) ORR 6/8 CR 4/8 PD on Rx 2/8 DLT 0/8 Dose Mods†: 8/8 | Zone 3/arm E (n = 5) (1 not evaluable) ORR 3/4 CR 2/4 PD on Rx 2/4 DLT 2/5 Dose Mods†: 2/5 | Zone 4/arm F (n = 0) |
| 200 (week 3+) 200 (week 2) 100 (week 1) | Zone 1/arm A (n = 2) ORR 2/2 CR 1/2 PD on Rx 0/2 DLT 0/2 Dose Mods†: 2/2 | Zone 2/arm B (n = 16) ORR 15/16 CR 6/15* PD on Rx 2/16 DLT 1/16 Dose Mods†: 6/16 | Zone 3/arm D (n = 4) ORR 2/4 CR 1/4 PD on Rx 2/4 DLT 0/4 Dose Mods†: 3/4 | |
| All subjects 20-100 VEN (cycle 0) | IBR (week 1+) mg per day | |||
|---|---|---|---|---|
| 280 | 420 | 560 | ||
| VEN (mg per day) | 400 (week 3+) 200 (week 2) 100 (week 1) | Zone 2/arm C (n = 8) ORR 6/8 CR 4/8 PD on Rx 2/8 DLT 0/8 Dose Mods†: 8/8 | Zone 3/arm E (n = 5) (1 not evaluable) ORR 3/4 CR 2/4 PD on Rx 2/4 DLT 2/5 Dose Mods†: 2/5 | Zone 4/arm F (n = 0) |
| 200 (week 3+) 200 (week 2) 100 (week 1) | Zone 1/arm A (n = 2) ORR 2/2 CR 1/2 PD on Rx 0/2 DLT 0/2 Dose Mods†: 2/2 | Zone 2/arm B (n = 16) ORR 15/16 CR 6/15* PD on Rx 2/16 DLT 1/16 Dose Mods†: 6/16 | Zone 3/arm D (n = 4) ORR 2/4 CR 1/4 PD on Rx 2/4 DLT 0/4 Dose Mods†: 3/4 | |
One subject had a response but could not be evaluated for CR status.
Dose modifications or holds.
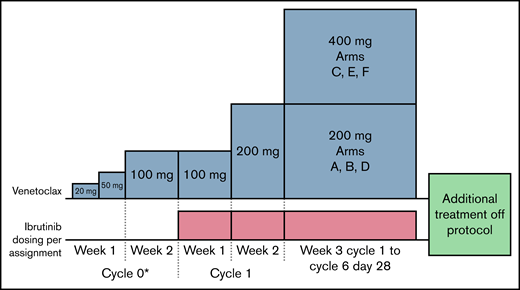
Treatment schema. Participants were initially treated with VEN, starting at 20 mg by mouth daily and increasing to 50 mg by mouth daily and 100 mg by mouth daily over 2 weeks (cycle 0*). Participants must have had at least 2 but no more than 4 days of 20 mg, and 3 but no more than 5 days of 50 mg before moving to 100 mg by mouth daily for 7 days. Ibrutinib (IBR), at the allocated dose, was added to venetoclax (VEN) at 100 mg by mouth daily for an additional 7 days (cycle 1, week 1). Weekly VEN dose titration was continued to the allocated dose as shown.
Treatment schema. Participants were initially treated with VEN, starting at 20 mg by mouth daily and increasing to 50 mg by mouth daily and 100 mg by mouth daily over 2 weeks (cycle 0*). Participants must have had at least 2 but no more than 4 days of 20 mg, and 3 but no more than 5 days of 50 mg before moving to 100 mg by mouth daily for 7 days. Ibrutinib (IBR), at the allocated dose, was added to venetoclax (VEN) at 100 mg by mouth daily for an additional 7 days (cycle 1, week 1). Weekly VEN dose titration was continued to the allocated dose as shown.
Study design, dose allocation, and statistical considerations
Details regarding the modeling approach and design considerations have been provided in a prior report.31 Briefly, participant enrollment to the study occurred in 2 allocation phases. In the first phase, each participant was sequentially allocated to 1 of 4 escalating zones comprising 6 dosing arms (Table 1). Once the DLT period elapsed without observance of a DLT in the first participant within each arm of a zone, subsequent participants would be allocated to the next increasing zone. Multiple participants could be enrolled in a given zone if the DLT observation period had not cleared. The second phase would begin if a DLT occurred at any time or at least 1 participant was enrolled in all arms and the DLT observation window had been cleared.
In the second allocation phase, eligible participants were allocated to a treatment arm based on the ORR at 2 months postcombination treatment start and DLT incidence in previously treated participants. Enrollment continued until 10 participants were allocated to an arm or the maximum enrollment was reached (23 patients). The optimal dose was defined as the arm with 10 participants assigned or that had an observed DLT rate ≤25% and maximized the ORR at 2 months.
Due to the potential safety and efficacy implications of the change in dosing strategy after study amendment related to the TLS event, enrollment was expanded and continued until 10 patients were allocated to the modified treatment arm or a total maximum study enrollment of 38 participants. The optimal dose was similarly defined but limited to those participants enrolled after the amendment. DLT and efficacy information was used from participants treated before the amendment to guide the starting arm for assignment after the amendment.
For the combination to be deemed promising, an observed ORR of at least 60% in 10 participants treated at the optimal dose combination by a 2-month disease assessment was needed as described previously.31 PFS and OS were estimated by the Kaplan-Meier method.
Safety and efficacy assessments
All participants who received any protocol treatment were monitored for adverse events (AEs), which were recorded based on CTCAE v4.03. DLTs were defined as AEs that were treatment-emergent and occurring during the beginning of VEN and IBR combination until day 28 of cycle 2 (56 days total). They included any grade ≥3 nonhematologic AE, febrile neutropenia of any duration, grade 4 thrombocytopenia or grade 3 thrombocytopenia with bleeding, grade 4 anemia, or any other AE that is thought to cause substantial morbidity for participants. Response was defined via Cheson et al32 and was evaluated by computer tomography (CT) every other cycle and positron emission tomography (PET)/CT at the end of treatment. For a response to be considered a CR, CT-based imaging had to be confirmed with a negative PET/CT and negative bone marrow (BM) biopsy if BM was involved at enrollment.
Trial conduct
The study was conducted at the University of Virginia in Charlottesville, VA, Washington University in St. Louis, MO, City of Hope in Duarte, CA, and Emory University in Atlanta, GA. Each institution’s institutional review board (IRB) approved the protocol, and the study was conducted according to general best practices and with adherence to the Declaration of Helsinki. All participants provided written informed consent. It was monitored and organized through the University of Virginia Clinical Trials Office and was governed by the FDA through IND number 124831 (CAP holder). Data cutoff was 12 October 2020.
Results
Patient characteristics
A total of 45 patients consented to the study from August 2015 to May 2019. Thirty-seven participants were deemed eligible (reasons for ineligibility included decreased renal function, prolonged QTc, and absence of measurable disease). Thirty-five participants were ultimately treated. A total of 17 eligible participants were enrolled before the dosing change prompted by the TLS event, and 20 eligible participants were treated post change. Two eligible participants were allocated to an arm at the time of the TLS event but had not started treatment and were removed from protocol due to study suspension for safety. Baseline characteristics are listed in Table 2, and baseline characteristics pre and postamendment are in supplement table 2.
Patient characteristics at enrollment
| Characteristic | Total (%) (n = 35) | A IBR 280 VEN 200 (n = 2) | B IBR 420 VEN 200 (n = 16) | C IBR 280 VEN 400 (n = 8) | D IBR 560 VEN 200 (n = 4) | E IBR 420 VEN 400 (n = 5) |
|---|---|---|---|---|---|---|
| Mean age in years (min-max) | 63 (49-82) | 75 (69-82) | 62 (54-76) | 66 (49-79) | 62 (53-68) | 56 (50-60) |
| % male | 29 (83%) | 2 (100%) | 14 (87%) | 6 (75%) | 3 (75%) | 4 (80%) |
| Prior auto transplant (yes), % | 15 (43%) | 0 (0%) | 8 (50%) | 4 (50%) | 1 (25%) | 2 (40%) |
| Refractory to last line of therapy (yes), % | 18 (51%) | 2 (100%) | 5 (31%) | 4 (50%) | 3 (75%) | 4 (80%) |
| Lines of prior Rx | ||||||
| 1 line | 20 (57%) | 1 (50%) | 12 (75%) | 3 (38%) | 3 (75%) | 1 (20%) |
| 2 lines | 13 (37%) | 1 (50%) | 4 (25%) | 3 (38%) | 1 (25%) | 4 (80%) |
| 3 lines | 2 (6%) | 0 (0%) | 0 (0%) | 2 (25%) | 0 (0%) | 0 (0%) |
| Stage at enrollment | ||||||
| 1-2 | 2 (6%) | 0 (0%) | 1 (6%) | 0 (0%) | 1 (25%) | 0 (0%) |
| 3 | 6 (17%) | 1 (50%) | 2 (13%) | 1 (13%) | 1 (25%) | 1 (20%) |
| 4 | 27 (77%) | 1 (50%) | 13 (81%) | 7 (88%) | 2 (50%) | 4 (80%) |
| MIPI at enrollment | ||||||
| Low | 17 (49%) | 1 (50%) | 9 (56%) | 2 (25%) | 2 (50%) | 3 (60%) |
| Intermediate | 12 (34%) | 1 (50%) | 5 (31%) | 2 (25%) | 2 (50%) | 2 (40%) |
| High | 6 (17%) | 0 (0%) | 2 (13%) | 4 (50%) | 0 (0%) | 0 (0%) |
| Characteristic | Total (%) (n = 35) | A IBR 280 VEN 200 (n = 2) | B IBR 420 VEN 200 (n = 16) | C IBR 280 VEN 400 (n = 8) | D IBR 560 VEN 200 (n = 4) | E IBR 420 VEN 400 (n = 5) |
|---|---|---|---|---|---|---|
| Mean age in years (min-max) | 63 (49-82) | 75 (69-82) | 62 (54-76) | 66 (49-79) | 62 (53-68) | 56 (50-60) |
| % male | 29 (83%) | 2 (100%) | 14 (87%) | 6 (75%) | 3 (75%) | 4 (80%) |
| Prior auto transplant (yes), % | 15 (43%) | 0 (0%) | 8 (50%) | 4 (50%) | 1 (25%) | 2 (40%) |
| Refractory to last line of therapy (yes), % | 18 (51%) | 2 (100%) | 5 (31%) | 4 (50%) | 3 (75%) | 4 (80%) |
| Lines of prior Rx | ||||||
| 1 line | 20 (57%) | 1 (50%) | 12 (75%) | 3 (38%) | 3 (75%) | 1 (20%) |
| 2 lines | 13 (37%) | 1 (50%) | 4 (25%) | 3 (38%) | 1 (25%) | 4 (80%) |
| 3 lines | 2 (6%) | 0 (0%) | 0 (0%) | 2 (25%) | 0 (0%) | 0 (0%) |
| Stage at enrollment | ||||||
| 1-2 | 2 (6%) | 0 (0%) | 1 (6%) | 0 (0%) | 1 (25%) | 0 (0%) |
| 3 | 6 (17%) | 1 (50%) | 2 (13%) | 1 (13%) | 1 (25%) | 1 (20%) |
| 4 | 27 (77%) | 1 (50%) | 13 (81%) | 7 (88%) | 2 (50%) | 4 (80%) |
| MIPI at enrollment | ||||||
| Low | 17 (49%) | 1 (50%) | 9 (56%) | 2 (25%) | 2 (50%) | 3 (60%) |
| Intermediate | 12 (34%) | 1 (50%) | 5 (31%) | 2 (25%) | 2 (50%) | 2 (40%) |
| High | 6 (17%) | 0 (0%) | 2 (13%) | 4 (50%) | 0 (0%) | 0 (0%) |
Cumulative toxicities by grade and arm
| Toxicity description | Total n = 35 | A IBR 280 VEN 200 (n = 2) | B IBR 420 VEN 200 (n = 16) | C IBR 280 VEN 400 (n = 8) | D IBR 560 VEN 200 (n = 4) | E IBR 420 VEN 400 (n = 5) | |||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Grade | 1 | 2 | 3 | 4 | 1 | 2 | 3 | 4 | 1 | 2 | 3 | 4 | 1 | 2 | 3 | 4 | 1 | 2 | 3 | 4 | 1 | 2 | 3 | 4 | |
| Hematologic | Febrile neutropenia | 1 | 1 | ||||||||||||||||||||||
| Bleeding/bruising | 3 | 2 | 1 | ||||||||||||||||||||||
| Neutropenia | 3 | 8 | 4 | 1 | 3 | 3 | 1 | 3 | 2 | 1 | 1 | ||||||||||||||
| Thrombocytopenia | 3 | 3 | 2 | 1 | 1 | 2 | 1 | 1 | 2 | 1 | 1 | ||||||||||||||
| Anemia | 4 | 2 | 1 | 2 | 1 | 1 | 1 | ||||||||||||||||||
| Gastrointestinal | Abd pain/bloating | 4 | 1 | 2 | 1 | ||||||||||||||||||||
| Diarrhea | 14 | 1 | 1 | 7 | 1 | 4 | 2 | 1 | 1 | ||||||||||||||||
| Constipation | 1 | 1 | |||||||||||||||||||||||
| Nausea | 11 | 5 | 3 | 1 | 2 | ||||||||||||||||||||
| Vomiting | 3 | 1 | 1 | 1 | |||||||||||||||||||||
| Dry mouth/dysgeusia | 7 | 6 | 1 | ||||||||||||||||||||||
| AST increase | 4 | 1 | 2 | 1 | 1 | 1 | |||||||||||||||||||
| ALT increase | 2 | 1 | 1 | 1 | 1 | ||||||||||||||||||||
| Cardiac | Hypertension | 2 | 2 | ||||||||||||||||||||||
| Atrial fib/flutter | 1 | 2 | 1 | 1 | 1 | ||||||||||||||||||||
| Infection | Lung infection | 1 | 1 | ||||||||||||||||||||||
| Upper respiratory | 10 | 2 | 2 | 5 | 1 | 2 | 2 | ||||||||||||||||||
| Skin infection | 1 | 1 | |||||||||||||||||||||||
| General | Edema | 3 | 1 | 2 | 1 | 1 | |||||||||||||||||||
| Fatigue | 5 | 4 | 1 | 1 | 2 | 2 | 2 | 1 | |||||||||||||||||
| Arthralgia/myalgia | 5 | 1 | 3 | 1 | 1 | 1 | |||||||||||||||||||
| Rash | 6 | 1 | 4 | 1 | |||||||||||||||||||||
| Toxicity description | Total n = 35 | A IBR 280 VEN 200 (n = 2) | B IBR 420 VEN 200 (n = 16) | C IBR 280 VEN 400 (n = 8) | D IBR 560 VEN 200 (n = 4) | E IBR 420 VEN 400 (n = 5) | |||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Grade | 1 | 2 | 3 | 4 | 1 | 2 | 3 | 4 | 1 | 2 | 3 | 4 | 1 | 2 | 3 | 4 | 1 | 2 | 3 | 4 | 1 | 2 | 3 | 4 | |
| Hematologic | Febrile neutropenia | 1 | 1 | ||||||||||||||||||||||
| Bleeding/bruising | 3 | 2 | 1 | ||||||||||||||||||||||
| Neutropenia | 3 | 8 | 4 | 1 | 3 | 3 | 1 | 3 | 2 | 1 | 1 | ||||||||||||||
| Thrombocytopenia | 3 | 3 | 2 | 1 | 1 | 2 | 1 | 1 | 2 | 1 | 1 | ||||||||||||||
| Anemia | 4 | 2 | 1 | 2 | 1 | 1 | 1 | ||||||||||||||||||
| Gastrointestinal | Abd pain/bloating | 4 | 1 | 2 | 1 | ||||||||||||||||||||
| Diarrhea | 14 | 1 | 1 | 7 | 1 | 4 | 2 | 1 | 1 | ||||||||||||||||
| Constipation | 1 | 1 | |||||||||||||||||||||||
| Nausea | 11 | 5 | 3 | 1 | 2 | ||||||||||||||||||||
| Vomiting | 3 | 1 | 1 | 1 | |||||||||||||||||||||
| Dry mouth/dysgeusia | 7 | 6 | 1 | ||||||||||||||||||||||
| AST increase | 4 | 1 | 2 | 1 | 1 | 1 | |||||||||||||||||||
| ALT increase | 2 | 1 | 1 | 1 | 1 | ||||||||||||||||||||
| Cardiac | Hypertension | 2 | 2 | ||||||||||||||||||||||
| Atrial fib/flutter | 1 | 2 | 1 | 1 | 1 | ||||||||||||||||||||
| Infection | Lung infection | 1 | 1 | ||||||||||||||||||||||
| Upper respiratory | 10 | 2 | 2 | 5 | 1 | 2 | 2 | ||||||||||||||||||
| Skin infection | 1 | 1 | |||||||||||||||||||||||
| General | Edema | 3 | 1 | 2 | 1 | 1 | |||||||||||||||||||
| Fatigue | 5 | 4 | 1 | 1 | 2 | 2 | 2 | 1 | |||||||||||||||||
| Arthralgia/myalgia | 5 | 1 | 3 | 1 | 1 | 1 | |||||||||||||||||||
| Rash | 6 | 1 | 4 | 1 | |||||||||||||||||||||
Optimal dose finding
A total of 7 participants were enrolled in the first allocation phase of the study when a DLT of prolonged grade 4 neutropenia occurred in arm E (VEN 400, IBR 420), prompting the initiation of the second phase. As a result, arm F (VEN 400, IBR 560) was not tested in the study's initial phase. An additional 10 participants were allocated via CRM before the TLS event (2 participants were allocated but not treated). Based on the prior 15 participants who received study therapy prior to the amendment, arm C (VEN 400, IBR 280) was considered a starting point after the amendment. After the amendment, allocation via CRM continued until 10 participants were allocated to arm B (VEN 200, IBR 420) and had DLT and response information available. While DLT and ORR information was maturing, 3 additional eligible participants were allocated for a total of 13 participants in arm B and an overall total of 20 participants treated after the amendment. The CRM did not recommend a participant be allocated to arm F (VEN 400, IBR 560). Arm B (VEN 200, IBR 420) was considered the optimal dose combination with an ORR of 15/16 (93.8%; 95% CI: 73.6% to 99.7%) and DLT incidence of 1/16 (6.2%; 95% CI: 0.3% to 26.4%). The trial conduct of the CRM allocation is represented in supplement table 3.
Treatment duration, toxicity, and response
Of the 35 participants allocated to and treated in an arm, 25 (71.4%) completed the 6 cycles of the combination treatment, 7 (17.1%) had disease progression or relapse before finishing therapy, and 3 (8.6%) had an AE causing treatment discontinuation. All participants were able to transition from cycle 0 (VEN only) to cycle 1 (IBR and VEN). Common AEs are summarized in Table 3. There were 3 DLTs consisting of persistent grade 4 neutropenia (arm E), persistent grade 3 diarrhea (arm E), and grade 3 respiratory disorder (arm B). Dose modifications on the study were allowed and are summarized in supplement table 4. As previously reported, the participant with the TLS event was able to continue study treatment and finished all 6 cycles of therapy.30 No other TLS events occurred.
Responses per arm are summarized in Table 1. One participant discontinued study treatment due to AE before disease assessment and was not evaluable for response; another participant had missing data at the end of treatment and was not evaluable for a complete response. Overall response for all arms was 82.3% (28/34; 95% CI: 65.5% to 93.2%) with a CR rate of 42.4% (14/33; 95% CI: 25.5% to 60.8%). Eight participants (23.5%) progressed during therapy; 3 of these had previously had a partial response. Notably, no demonstrable difference in response or progression was noted between the arms.
For the 25 participants who finished the study treatment after 6 months, 14 (56%) continued to take IBR (9 at a dose of 560 mg by mouth daily and 5 at lower doses), and 11 (44%) had other therapies (8 unknown and 3 chemotherapy).
PFS and OS
PFS and OS were estimated from cycle 1, day 1 (not including cycle 0; there were no PFS or OS events in cycle 0). The median follow-up for all alive patients was 26.7 months (5.5 months to 53.4 months). Median PFS and OS for the entire cohort were estimated to be 10.7 and 28.3 months, respectively. Median PFS and OS for arm B, the optimal dose combination, were not reached with a median follow-up of 22.9 months (5.6 months to 53.4 months) (Figure 2). Survival should be interpreted with caution given the study treatment only lasted for 6 cycles. In those who finished 6 cycles of combination therapy, there was an improvement in PFS for those that continued IBR over other treatments; however, the limited numbers did not reach statistical significance (P = .10) (Figure 3). Individual patient outcomes are summarized in a swimmer plot in supplemental Figure 1.
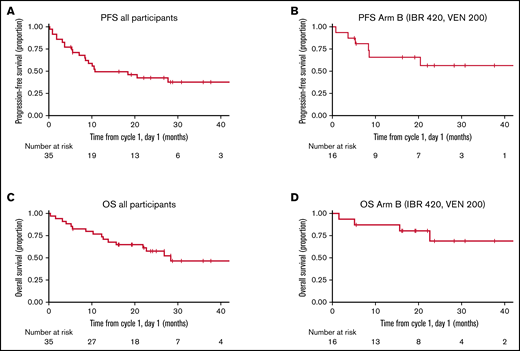
PFS and OS. (A) PFS for all participants. (B) PFS for the optimal dose, arm B (IBR 420 mg daily and VEN 200 mg daily). (C) OS for all participants. (D) OS for the optimal dose, arm B (IBR 420 mg daily and VEN 200 mg daily).
PFS and OS. (A) PFS for all participants. (B) PFS for the optimal dose, arm B (IBR 420 mg daily and VEN 200 mg daily). (C) OS for all participants. (D) OS for the optimal dose, arm B (IBR 420 mg daily and VEN 200 mg daily).
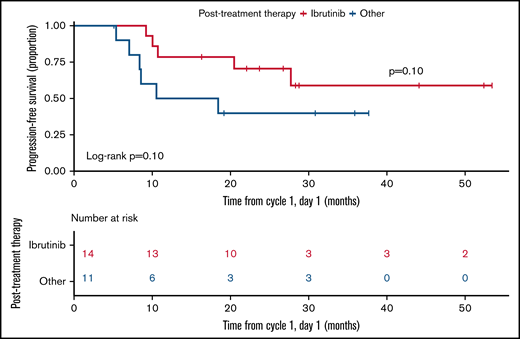
PFS in those finishing study therapy. PFS starts at the end of therapy and compares those that were treated with IBR (red) to those that were not (blue).
PFS in those finishing study therapy. PFS starts at the end of therapy and compares those that were treated with IBR (red) to those that were not (blue).
Discussion
We present the only known phase 1 dose-finding study of IBR and VEN in NHL that evaluates the dosing of both drugs. Prior clinical studies28,29 of the IBR and VEN combination have used the single-agent approved doses in combination as phase 2 studies or have attempted to increase the approved doses in the phase 1 study. Our approach was novel in finding the optimal dose instead of the maximum tolerated dose, utilizing a CRM to allocate participants based on prior participants’ efficacy and toxicity. Our primary finding shows that the optimal dose was arm B (IBR 420 mg by mouth daily, VEN 200 mg by mouth daily), which is lower than other studies are using (SYMPATICO study, NCT03112174)33,34 or have used (AIM or OAsIs study)28,29,35 in relapsed MCL. The SYMPATICO,33,34 AIM,28,35 and OAsIs29 studies use IBR 560 mg by mouth daily and VEN 400 mg by mouth daily in MCL, a dose combination that we were not able to test due to DLTs and adequate response at lower dose levels. The reports of these studies33-35 have reported frequent dose reductions in upwards of 60% of patients. The OAsIs study had a dose-finding portion of the study (evaluating higher doses of VEN) with limited DLTs; however, our definition of DLTs was conservative, and our DLT interval was long (8 weeks). Thus, the definition of DLT between the 2 studies is different, the current study being more cautious and accounting for the difference in DLT incidence. Toxicity has been reported with VEN doses higher than 400 mg when combined with IBR, including frequent hematologic toxicities.29,36 Given these overall findings, there is comfort that dose reductions of the combination on these studies can be effective and safe.
The ORR of 83% and a CR rate of 42% (ORR of 93.7% and a CR rate of 40% for the optimal arm B) compares favorably to single-agent IBR ORR of 66% and CR rate 20%16 and is comparable to the ORR of 67% in the AIM study28 and 70% in the OAsIs study (relapsed, combination cohort only).29 There does appear to be an improvement in CR rate in the prior studies, likely related to prolonged therapy on these studies, the use of obinutuzumab in the OAsIs study, and our strict definition of CR rate (PET negative and BM negative) by Lugano classification.32 Thus, the combination is worthy of further study, as is currently underway in the SYMPATICO phase 3 study of IBR+VEN vs IBR+placebo in MCL (NCT03112174). Duration of response and survival are difficult to compare as our study only provided the combination for six 28-day cycles, and subsequent therapies were left to investigator discretion. This also makes the achievement of minimal residual disease negative status, which we were not able to perform, difficult to interpret. The attribution of subsequent therapies to prolonged outcomes is uncertain, though there was a trend for improvements for those who continued IBR monotherapy.
There are limitations to our study. The first would be that our clinical parameters at entry would portend for a more favorable MCL outcome (Table 2). This limitation is likely due to excluding participants deemed to be at high risk for TLS and prior BTKi exposure. The latter was significant at a time of enrollment where BTKi was mainly used in the second-line setting, thus prioritizing enrollment to those who had relapsed after initial treatment. Second, we do not have robust information regarding known biologic determinants of MCL such as Ki67, TP53 mutations or alterations, or blastic morphology, as the central pathology review was not performed and biopsy was not required at study entry. Third, the trial conduct of a CRM does not allow for matching of the various arms by clinical characteristics; therefore, there are differences in the arms. Thus, there is a risk that arm B was identified as optimal based on clinical differences. However, the totality of the evidence in this study would suggest that combination doses lower than the single-agent dose of both IBR and VEN are safe and effective. Finally, our decision to limit the combination to six 28-day cycles may, in retrospect, be too short of a course. At the time of trial design, the intention was to use the combination as a 6-month intensive therapy followed by maintenance therapy of the investigator's discretion (single-agent IBR was recommended); therefore, survival (PFS and OS) are difficult to interpret. However, this study was not designed to evaluate the long-term benefit of this combination but rather to systematically assess the optimal dose when both agents are used in combination.
By starting treatment with VEN for all participants, we provide some insight into the single-agent use of VEN in MCL. First, as has been reported,30 an initial dose of 100 mg did cause TLS, even when limiting patients to low or medium tumor burden, and VEN must be given with caution in MCL. While we did not test for response after only 1 to 2 weeks of treatment with VEN alone, we anecdotally saw rapid clinical improvements in disease burden for some patients treated at the 100 mg daily dose. After the amendment, changing the dosing strategy to start at 20 mg of VEN with a ramp-up to 50 mg in 1 week and then to 100 mg over an additional week, we did not see any additional TLS events or any significant impact on response rate at lower doses. Thus, a more rapid dose escalation than is used in CLL26 can be used in MCL if monitoring carefully for TLS and limiting patients to low to medium risk for TLS. It is often important to reach an effective treatment dose with MCL as active disease can be clinically significant and require rapid treatment. A dose-escalation starting at 20 mg and reaching a dose of 100 mg by 1 week can allow for this if carefully monitored. By using VEN before IBR, we were able to get to the maximum dose of both drugs by 4 weeks, achieving an effective dose more quickly than the OAsIs29 and AIM28 studies. This also avoided the concern for TLS after peripheralization of leukemic MCL seen with IBR.
There was no major response advantage for higher doses of the combination observed in this study. Given our dosing strategy, we can only adequately discuss progressions occurring in the first 6 months of treatment. Nonresponse or progression following a response does not seem to be related to the dose of the combination. This suggests underlying biology driving early resistance to the combination of IBR and VEN that we would hypothesize would not be adequately salvaged with higher doses of either drug. Dosing of IBR has been reevaluated in the literature recently37,38 and has been suggested that, at least in CLL, attenuated doses can still provide disease control. Mechanisms of resistance with the combination have not been fully identified; however, some investigators have identified potential targets. In the AIM study, the SWI-SNF mutations seemed to mediate resistance through the upregulation of Bcl-xL.39 In preclinical models, we have found that the tumor microenvironment modulation can evoke IBR and VEN resistance through NF-kB-mediated upregulation of antiapoptotic proteins.40 With ongoing investigation, we plan to utilize clinical specimens collected during this study to evaluate these biologic determinants predicting response and occurring at progression.
Finally, we elected to proceed with a dose-finding study at the inception of this trial as both IBR and VEN were known to be metabolized through CYP3A. Further, it was known that BTK occupancy was full and robust at the FDA-approved dose of 560 mg by mouth daily, and even lower doses had full occupancy.15 Thus, the 2 drugs' interaction was hypothesized to increase effective drug levels that may allow for a lower dose without negative effects on efficacy. Our finding of an optimal combination of IBR and VEN that is lower than both drugs used as single agents would support this hypothesis. Targeted small molecule inhibitors are increasingly being combined in clinical practice, and it is important to recognize drug interactions as doses used as a single agent may cause toxicities when used in combination. Our study would suggest that IBR 420 mg by mouth daily and VEN 200 mg by mouth daily has a similar response rate to other studies of the combination. At this dose, there were fewer dose interruptions or reductions than previously reported28,33,35 and improved toxicity profile.
Acknowledgments
The authors would like to thank the patients, family, and staff that helped make this study feasible. They acknowledge the work of the UVA Clinical Trials Office, Office of Cancer Research, Grants and Contracts, and Biorepository and Tissue Research Facility in the execution and conduct of the study. This project was developed as part of the Lymphoma Research Foundation’s Lymphoma Clinical Research Mentoring Program (C.A.P.). This publication was supported by the University of Virginia Cancer Center (P30CA044579).
This work was supported by a grant from AbbVie Inc. to the University of Virginia. The design, execution, results, and findings were made by the authors independent from AbbVie Inc. The findings were reviewed by AbbVie prior to submission for publication.
Authorship
Contribution: C.A.P. designed the research, performed the research, analyzed the data, contributed patient data, and wrote the paper; N.A.W. designed the research, monitored and analyzed the data and edited the paper; B.S.K. designed the research, contributed patient data, and edited the paper; L.E.B., R.W.C., and J.B.C. contributed patient data and edited the paper; N.E.V. monitored and anaylazed the data;
G.R.P. designed the research, monitored and analyzed the data, and edited the paper; and M.E.W. designed the research, contributed patient data, and edited the paper.
Conflict-of-interest disclosure: C.A.P.: Consulting: BeiGene, Genentech, Epizyme, Kite/Gilead, Morphosys, Pharmacyclics, and Jansen; Research funding: BeiGene, Kite/Gilead, TG Therapeutics, Acerta, Infinity/Veristem, AbbVie, and Velosbio/Merck. B.S.K. Consulting: Abbvie, Pharmacyclics, Janssen, and AstraZeneca; Research funding: Abbvie, AstraZeneca, Acerta, BeiGene. J.B.C.: Consulting: Janssen, AstraZeneca, Beigene, Kite/Gilead, Adaptive, Cellectar, Loxo, Aptitude Health; Research: AstraZeneca, Loxo, Genentech, Novartis, BMS/Celgene, Lam Therapeutics, BioInvert, Atara. M.E.W.: Consulting: Abbvie, Janssen; Honorarium: Xian, Janssen; Research funding: Janssen, Pharmacyclics. The remaining authors declare no competing financial interests.
Correspondence: Craig A. Portell, Univeristy of Virginia, Division of Hematology/Oncology, PO Box 800716, Charlottesville, VA 22908-0716; e-mail: cp4ys@hscmail.mcc.virginia.edu.

