

Key Points
Momelotinib shows high efficacy against FLT3-mutated AML cells, including quizartinib-resistant activation loop variants.
Momelotinib effectively suppresses intrinsic resistance conferred by FLT3 ligand and hematopoietic cytokines (GM-CSF and IL-3).

Abstract
Despite the introduction of more selective FLT3 inhibitors to treat FLT3-mutated acute myeloid leukemia (AML), remissions are short lived, and patients show progressive disease after an initial response. Acquisition of resistance-conferring genetic mutations and growth factor signaling are 2 principal mechanisms that drive relapse. FLT3 inhibitors targeting both escape mechanisms could lead to a more profound and lasting clinical response. Here, we show that the JAK2 inhibitor momelotinib is an equipotent type 1 FLT3 inhibitor. Momelotinib showed potent inhibition of FLT3-internal tandem duplication in mouse and human primary cells and effectively suppressed its clinically relevant resistant variants within the activation loop at residues D835, D839, and Y842. Additionally, momelotinib efficiently suppressed the resistance mediated by growth factors and hematopoietic cytokine-activated JAK2 signaling. Consequently, concomitant inhibition of FLT3 and suppression of growth factor signaling by momelotinib treatment showed better efficacy in suppressing leukemia in a preclinical murine model of AML. Altogether, these data provide evidence that momelotinib is an effective type 1 dual JAK2/FLT3 inhibitor and may offer an alternative to gilteritinib. Its ability to impede the resistance conferred by growth factor signaling and activation loop mutants suggests that momelotinib treatment could provide a deeper and durable response and, thus, warrants its clinical evaluation.
Introduction
Approximately one third of patients with acute myeloid leukemia (AML) harbor internal tandem duplication (ITD) of the juxtamembrane region of FLT3, which is associated with poor treatment outcome and overall survival, even after stem cell transplantation.1,2 The clinical efficacy of first-generation Flt3 inhibitors was marred because of poor affinity and bioavailability. Second-generation FLT3 inhibitors designed to overcome previous shortcomings have shown superior response to their predecessors but failed to induce a durable response.3 Acquisition of drug-resistant mutations in FLT3 kinase or activation of alternate signaling pathways are commonly observed in relapsed patients. For instance, quizartinib, which binds to the inactive conformation, is prone to select kinase-activating mutations from the gatekeeper and activation loop residues.4,5 Therefore, patients harboring FLT3-TKD mutations (D835) are nonresponsive to quizartinib. As noted earlier with imatinib resistance in chronic myeloid leukemia,6,7 FLT3 inhibitors targeting the active conformation (gilteritinib and crenolanib) effectively suppressed the kinase-activating quizartinib-resistant mutations,8 but they are prone to select resistant clones harboring mutations in genes supporting survival, such as RAS, PTPN11, TET2, and IDH1/2.9-13 In addition, a significant fraction of leukemic cells residing in the bone marrow is refractory to tyrosine kinase inhibitors (TKIs), which manifests as minimal residual disease whose eradication is necessary for a durable response. Activation of JAK2 and MAPK pathways by growth factor signaling, such as FLT3 ligand,14 chemokines, and cytokines (CXCR4, FGF2,15 interleukin-3 [IL-3], and granulocyte-macrophage colony-stimulating factor [GM-CSF]16 ), was reported to drive TKI refractoriness. Moreover, myelosuppression and cardiotoxicity due to off-target inhibition of c-KIT and hERG by FLT3 inhibitors pose additional clinical challenges.
FLT3 inhibitors active against its resistant variants and suppressing the cytokine and chemokine resistance while lacking selectivity for c-KIT and hERG may provide a better therapeutic outcome. Here, we show that momelotinib, a JAK2 inhibitor,17 is an equipotent type I FLT3 inhibitor and does not inhibit c-KIT. Like gilteritinib, it selectively binds to the active conformation; therefore, it potently inhibits the resistance conferred by FLT3-activating mutations from the activation loop. Unlike gilteritinib, patients treated with momelotinib did not show QT prolongation and cardiotoxicity, suggesting that it does not inhibit hERG and c-KIT.18-20 Altogether, our preclinical data support its evaluation in FLT3-mutated AML.
Methods
Plasmids and inhibitors
pMSCV-Ires-GFP, pMSCV-FLT3WT-Ires-GFP, and pMSCV-FLT3ITD-Ires-GFP were described previously.21 Ruxolitinib and pacritinib were purchased from Chemietech Inc. Quizartinib, sorafenib, midostaurin, lestaurtinib, crenolanib, gilteritinib, and momelotinib were purchased from AdooQ Bioscience.
Cell lines
BaF3 cells stably expressing quizartinib-resistant variants were generated as described earlier.21 Human AML cell lines K562, Molm13, and MV4-11 were described previously.22
Cell viability assays, western blotting, molecular docking, and in vivo drug treatment analysis were performed as described earlier. A detailed protocol is provided in supplemental Methods.
Results and Discussion
While studying the efficacy of combinatorial targeting of JAK2 and FLT3 using ruxolitinib or momelotinib (as JAK2 inhibitors) with quizartinib (as an Flt3 inhibitor) on AML cells, we observed inhibitory activity of momelotinib on FLT3ITD cells. Unlike ruxolitinib, momelotinib alone can efficiently suppress the proliferation of BaF3-JAK2V617F (50% inhibitory concentration [IC50], 325 nM), BaF3-FLT3-WT (IC50, 320 nM), and BaF3-FLT3ITD (IC50, 401 nM) cells (Figure 1A; supplemental Figure 1A-C). Similarly, like quizartinib and gilteritinib, momelotinib inhibited the proliferation of patient-derived FLT3ITD AML cells, with IC50’s of 165 to 203 nM, while lacking activity against non-FLT3–mutated AML cells (Figure 1B; supplemental Figure 1D-F). Likewise, momelotinib led to a dose-dependent biochemical inhibition of phosphorylated (p)FLT3, pSTAT5, and pERK1/2 in FLT3ITD-expressing mouse and human cells, paralleling the inhibitory concentrations of cellular proliferation (Figure 1C-D; supplemental Figure 1G-H). In contrast, FLT3-mutated and non-FLT3–mutated AML cell lines were resistant to ruxolitinib, with the exception of TF-1 (dependent on Jak2 signaling because it requires IL-3 for survival), and it failed to inhibit pFLT3 and pSTAT5, suggesting that the inhibition of FLT3 signaling in AML is independent of JAK2 inhibition (supplemental Figure 1I-K). To determine the on-target inhibition of FLT3, we performed a comparative analysis of momelotinib with other FLT3 inhibitors against resistant variants of FLT3ITD. All quizartinib-resistant FLT3 mutants conferred resistant to sorafenib and pacritinib (supplemental Figure 2). In contrast, momelotinib, like type I FLT3 inhibitors (gilteritinib, midostaurin, lestaurtinib, and crenolanib), potently inhibited the FLT3ITD-resistant variants from the activation loop (D835Y, D839G, and Y842H) but failed to curb the gatekeeper variant F691L (Figure 1E-G; supplemental Figure 2). Accordingly, momelotinib inhibited the FLT3 and STAT5 phosphorylation at concentrations paralleling the inhibitory concentrations required for cell proliferation (Figure 1H). Next, we determined the inhibitory activity of momelotinib on FLT3ITD compound mutations (gatekeeper mutation F691L with activation loop variant D835Y or Y842H on the same allele). Surprisingly, we observed higher momelotinib sensitivity (fourfold) for the compound mutant FLT3ITD/F691L/Y842H (IC50, 1001 nM) in comparison with FLT3ITD/F691L or FLT3ITD/F691L/D835Y (Figure 1E), suggesting that activation loop mutants might break the resistance conferred by the gatekeeper mutation by altering the conformational dynamics, as described earlier.6,7,23 It also implies that the gatekeeper mutant F691L might be stabilizing a unique conformation to prevent momelotinib binding, rather than a steric hindrance observed with quizartinib and gilteritinib.
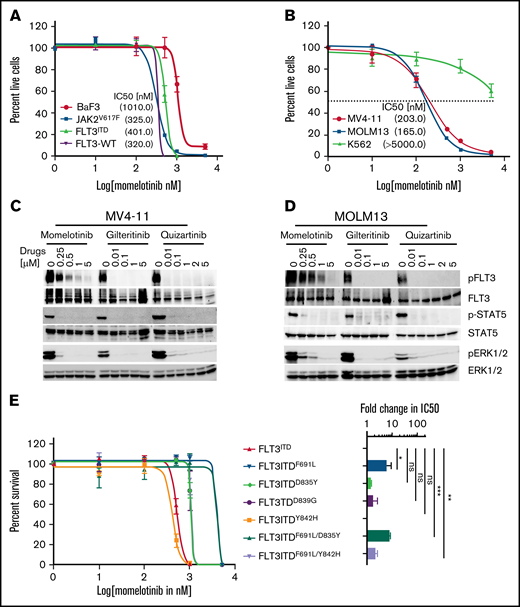
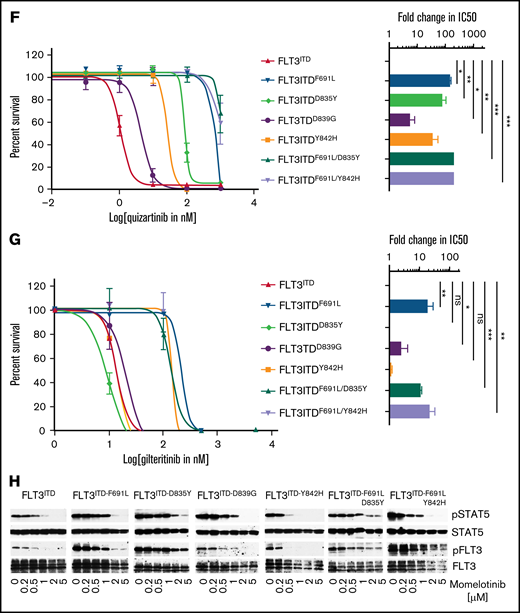
Momelotinib potently inhibits FLT3ITD and its resistant variants from the activation loop. (A) Sigmoidal curve showing the viability of BaF3 cells expressing FLT3-WT, Jak2V617F, and FLT3ITD treated with dimethyl sulfoxide or increasing concentrations of momelotinib for 72 hours. IC50 values for each cell line is indicated in parentheses. (B) Sigmoidal curve showing the potent inhibition of human FLT3ITD-mutant AML cells (MV4-11 and MOLM13), whereas proliferation of K562 cells was not significantly affected. IC50 values for each cell line is indicated in parentheses. pFLT3 and pSTAT5 levels determined by western blotting using total cell extracts of MV4-11 cells (C) and MOLM13 cells (D) treated with increasing concentrations of momelotinib for 2 hours. Dose-response sigmoidal curve showing the proliferation of BaF3 cells expressing FLT3ITD and its quizartinib-resistant variants at different concentrations of momelotinib (E), quizartinib (F), and gilteritinib (G) (left panels). Fold differences in the IC50 values for each FLT3ITD variant normalized to FLT3ITD are presented as a bar graph with a logarithmic scale (right panels). Note that momelotinib efficiently inhibited the activation loop quizartinib-resistant mutants, as well as compound mutant FLT3ITD/F691L/Y842H, which is fully resistant to gilteritinib (brown bar). (H) Immunoblot analysis showing inhibition of the kinase activity of FLT3ITD and resistant variants treated with different concentrations of momelotinib. Total cell extracts from the cells treated with momelotinib for 2 hours were probed with anti-pFLT3, anti-pSTAT5, anti-FLT3, and anti-STAT5 antibodies. Representative cell proliferation data (± standard deviation) are shown from 2 independent experiments. Error bars represent standard error of the mean. *P < .05, **P < .01, ***P < .001. ns, not significant.
Momelotinib potently inhibits FLT3ITD and its resistant variants from the activation loop. (A) Sigmoidal curve showing the viability of BaF3 cells expressing FLT3-WT, Jak2V617F, and FLT3ITD treated with dimethyl sulfoxide or increasing concentrations of momelotinib for 72 hours. IC50 values for each cell line is indicated in parentheses. (B) Sigmoidal curve showing the potent inhibition of human FLT3ITD-mutant AML cells (MV4-11 and MOLM13), whereas proliferation of K562 cells was not significantly affected. IC50 values for each cell line is indicated in parentheses. pFLT3 and pSTAT5 levels determined by western blotting using total cell extracts of MV4-11 cells (C) and MOLM13 cells (D) treated with increasing concentrations of momelotinib for 2 hours. Dose-response sigmoidal curve showing the proliferation of BaF3 cells expressing FLT3ITD and its quizartinib-resistant variants at different concentrations of momelotinib (E), quizartinib (F), and gilteritinib (G) (left panels). Fold differences in the IC50 values for each FLT3ITD variant normalized to FLT3ITD are presented as a bar graph with a logarithmic scale (right panels). Note that momelotinib efficiently inhibited the activation loop quizartinib-resistant mutants, as well as compound mutant FLT3ITD/F691L/Y842H, which is fully resistant to gilteritinib (brown bar). (H) Immunoblot analysis showing inhibition of the kinase activity of FLT3ITD and resistant variants treated with different concentrations of momelotinib. Total cell extracts from the cells treated with momelotinib for 2 hours were probed with anti-pFLT3, anti-pSTAT5, anti-FLT3, and anti-STAT5 antibodies. Representative cell proliferation data (± standard deviation) are shown from 2 independent experiments. Error bars represent standard error of the mean. *P < .05, **P < .01, ***P < .001. ns, not significant.
To understand the structural basis of momelotinib inhibition, we performed docking studies using FLT3 coordinates representing inactive (PDB: 4XUF) and active conformations (PDB: 5X02). Our docking analysis predicts that momelotinib, like gilteritinib, binds to only an open and enzymatically active conformation of FLT3, in which residue Phe830 of the DFG motif is displaced to accommodate the phenyl-cyanomethyl ring (Figure 2A-E). Momelotinib anchors to the ATP site by 3 hydrogen bonds coordinated by Cys694 from the hinge region and Glu817 and Ser618 from the P-loop (Figure 2B). The study also revealed that the inactive conformation is incompatible for momelotinib and gilteritinib binding because of the steric clash with Phe 830 (Figure 2C-D). Instead, it binds explicitly with an active conformation, DFG-in state (Figure 2E). Mutations in the activation loop will destabilize the inactive conformation and drive conformational state equilibrium toward the active state, which is preferentially targeted by type 1 inhibitors, explaining the enhanced momelotinib activity against activation loop mutants. Mutation at gatekeeper residue (F691) conferred resistance to gilteritinib and momelotinib. A leucine substitution for Phe691 has conferred resistance to type I and type II inhibitors; however, the extent of resistance is much greater for type II inhibitors, because gatekeeper mutation causes a direct steric hindrance to drug binding, as well as destabilizes the inactive conformation to which type II inhibitors preferentially bind (Figure 2C-E). We observed that gatekeeper mutant causes steric hindrance to gilteritinib but not to momelotinib. It is not clear how F691L confers resistance to momelotinib. Based on our model, we speculate that substituting Leu for Phe691 will weaken the hydrophobic spine, which may stabilize an intermediate conformation to which type I inhibitors seemingly have a lower affinity. Therefore, stabilizing the spine by strong kinase-activating mutations, such as Y842H, may restore inhibitor sensitivity. Our data support this notion, because a compound mutant, FLT3ITD/F691L/Y842H, showed enhanced sensitivity to momelotinib, as well as higher pFLT3 levels (Figure 1H; supplemental Figure 3). Similarly, F691L mutations conferred resistance to lestaurtinib (a type I inhibitor), but both compound mutations (F619L/D835Y and F619L/Y842H) show increased sensitivity compared with F691L alone (supplemental Figure 2E). Altogether, these data suggest that a combined F691L and Y842H mutation shifts the structural equilibrium to an active state conformation for which momelotinib has a greater affinity.
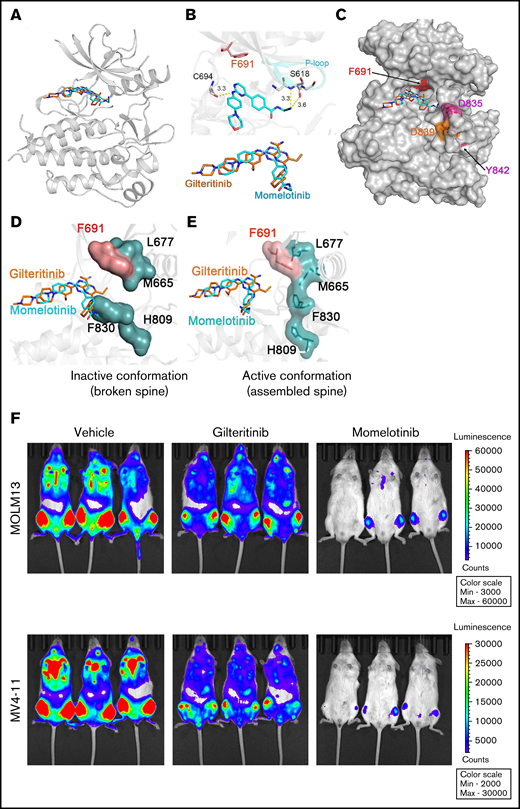
Momelotinib is a type I FLT3 inhibitor and effectively suppresses leukemic progression in mice. (A) Ribbon depiction of a structural model of FLT3 momelotinib was built using quizartinib (PDB: 4XUF) and gilteritinib (PDB: 6JQR) coordinates. A stick representation of momelotinib (cyan) and gilteritinib (brown) shows their binding to the ATP site. (B) Close-up view of the active site of the FLT3 kinase showing the interaction of momelotinib with Cys694 (C694) from the kinase hinge region and Ser618 (S618) from the P-loop through 3 hydrogen bonds (upper panel). Similar binding of gilteritinib and momelotinib (lower panel). (C) Surface depiction of FLT3 kinase with gilteritinib showing the binding of momelotinib and gilteritinib. The activation loop residues D835 (pink), D839 (brown), and Y842 (pink) are frequently mutated in patients treated with type II inhibitors. (D) Model of FLT3 inactive conformation docked with momelotinib and gilteritinib showing steric clash with Phe830 and broken hydrophobic spine. (E) Model of FLT3 active kinase with a stable hydrophobic spine showing the binding of momelotinib and gilteritinib, which provides an explanation why they favor active kinase conformation for binding. (F) Luminescence imaging of NSGS mice transplanted with MOLM13 (upper panels) and MV4-11 (lower panels) at 2 weeks posttransplantation. One million MOLM13-luciferase-Cherry cells (upper panels) and MV4-11-luciferase-Cherry cells (lower panels) were transplanted by tail vein injection into NSGS mice. Momelotinib treatment (100 mg/kg daily in phosphate-buffered saline by intraperitoneal injection) was started 2 days posttransplant. Control mice were injected with vehicle (phosphate-buffered saline).
Momelotinib is a type I FLT3 inhibitor and effectively suppresses leukemic progression in mice. (A) Ribbon depiction of a structural model of FLT3 momelotinib was built using quizartinib (PDB: 4XUF) and gilteritinib (PDB: 6JQR) coordinates. A stick representation of momelotinib (cyan) and gilteritinib (brown) shows their binding to the ATP site. (B) Close-up view of the active site of the FLT3 kinase showing the interaction of momelotinib with Cys694 (C694) from the kinase hinge region and Ser618 (S618) from the P-loop through 3 hydrogen bonds (upper panel). Similar binding of gilteritinib and momelotinib (lower panel). (C) Surface depiction of FLT3 kinase with gilteritinib showing the binding of momelotinib and gilteritinib. The activation loop residues D835 (pink), D839 (brown), and Y842 (pink) are frequently mutated in patients treated with type II inhibitors. (D) Model of FLT3 inactive conformation docked with momelotinib and gilteritinib showing steric clash with Phe830 and broken hydrophobic spine. (E) Model of FLT3 active kinase with a stable hydrophobic spine showing the binding of momelotinib and gilteritinib, which provides an explanation why they favor active kinase conformation for binding. (F) Luminescence imaging of NSGS mice transplanted with MOLM13 (upper panels) and MV4-11 (lower panels) at 2 weeks posttransplantation. One million MOLM13-luciferase-Cherry cells (upper panels) and MV4-11-luciferase-Cherry cells (lower panels) were transplanted by tail vein injection into NSGS mice. Momelotinib treatment (100 mg/kg daily in phosphate-buffered saline by intraperitoneal injection) was started 2 days posttransplant. Control mice were injected with vehicle (phosphate-buffered saline).
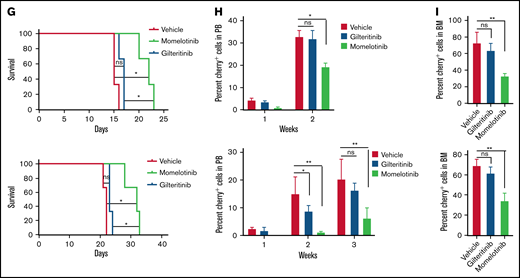
A recent study showed that activation of JAK2 by hematopoietic cytokines (GM-CSF and IL-3) promotes TKI refractoriness, which serves as a reservoir to develop secondary resistance. As a proof of concept, a combination of inhibitors (gilteritinib and ruxolitinib), targeting FLT3 and JAK2, respectively, showed a better in vivo response.16 Given that momelotinib inhibits FLT3 and JAK2, we analyzed the efficacy of momelotinib alone on leukemic cells grown with GM-CSF and IL-3. As expected, GM-CSF and IL-3 conferred resistance to quizartinib and gilteritinib, whereas momelotinib completely suppressed the cytokine-mediated resistance (supplemental Figure 4). These data provide evidence that momelotinib is a dual JAK2/FLT3 inhibitor, and it might be more effective in suppressing the disease as a single agent. As envisioned, momelotinib treatment in a preclinical mouse model of AML (NSGS mice expressing hematopoietic cytokines predicted to confer gilteritinib resistance) effectively suppressed the leukemic progression, whereas gilteritinib treatment was ineffective (Figure 2F-I). Because off-target inhibition of c-KIT by most FLT3 inhibitors causes myelosuppression, we compared the inhibitory activity of gilteritinib, quizartinib, and momelotinib using BaF3 cells expressing constitutively active c-KIT (D816Y) and its resistant variants (T681I). As shown in supplemental Figure 5, both known KIT inhibitors, dasatinib and quizartinib, inhibited c-KITD816Y but failed to inhibit the gatekeeper variant (KITD16Y/T681I), supporting their on-target activity. In contrast, momelotinib, like gilteritinib, did not inhibit either KIT mutant, suggesting that it lacks activity against c-KIT and, hence, will not be myelosuppressive.
Next, we evaluated the efficacy of momelotinib on primary human AML cells (supplemental Figure 6A). As expected, gilteritinib efficiently inhibited the proliferation of FLT3ITD AML cells (IC50, 0.5-0.6 nM) when assayed without resistance-conferring cytokines, but it lacked activity against non-FLT3–mutated and FLT3ITD cells harboring additional high-risk AML mutations (supplemental Figure 6B-C). In contrast, momelotinib inhibited the proliferation of FLT3ITD AML cells (IC50, 7-279 nM) and suppressed the growth factor–mediated resistance (supplemental Figure 6D-E). As expected, non-FLT3–mutated AML cells conferred resistance to momelotinib. Interestingly, the FLT3ITD-positive AML cell sample harboring additional mutations (AML3) that showed primary resistance to gilteritinib is effectively suppressed by momelotinib in both cytokine concoctions, with an elevated IC50 ∼ 270 nM. A recent pharmacokinetics and pharmacodynamics analysis revealed that only ∼19% (∼350 nM) of momelotinib is freely available, which, nonetheless, fully inhibited Jak2 with an IC50 ∼ 259 nM.33 To determine the in vivo efficacy of momelotinib on FLT3, a plasma-inhibitory assay was performed using MOLM13 cells and a primary patient sample (supplemental Figure 6F-G), which showed ∼90% inhibition of FLT3 activity at 340 nM of drug concentration in MOLM13 cells (supplemental Figure 6F, left panel). Interestingly, Flt3 activity in the primary patient sample was fully inhibited at 16 nM of drug concentration, which corresponds to a concentration that is 20 times lower than its Cmax value (supplemental Figure 6F, right panel). As expected, lestaurtinib failed to inhibit FLT3 in a similar assay, recapitulating the previous observation (supplemental Figure 6G). These data clearly show that momelotinib effectively suppresses FLT3 activity in vivo. Nonetheless, further studies are needed to fully characterize the effect of momelotinib in preclinical models of FLT3-mutated AML and to fully elucidate the mechanism of action.
Clinical efficacy of FLT3 TKIs is limited by the growth factor–mediated intrinsic resistance that drives the emergence of resistant clones and the off-target inhibition of c-KIT and hERG that result in myelosuppression and QT prolongation, respectively. Our preclinical studies show that momelotinib overcomes most of the hurdles faced by the FLT3 TKIs that are currently used for effective treatment outcomes. We show that momelotinib is a type 1 dual JAK2/FLT3 inhibitor that effectively suppresses the resistance mediated by activation loop mutants and growth factor signaling. Severe cases of myelosuppression are not reported in the clinical studies performed with momelotinib, which supports our finding that it lacks activity against c-KIT. Altogether, our findings indicate that momelotinib may provide a durable treatment response.
Acknowledgments
This work was supported by National Institutes of Health National Cancer Institutes grants R01 CA211594 and R01 CA250516 (M.A.). M.A. is a recipient of a Bridge Grant from the American Society of Hematology.
Authorship
Contribution: M. Azhar, M.K., Z.K, and M. Azam designed the experiments; M.A., M.K., and Z.K. performed experiments and analyzed data; D.S. provided critical reagents and supervised the data analyses; and M. Azhar, M.K., and M. Azam wrote the manuscript.
Conflict-of-interest disclosure: D.S. serves on the scientific advisory board for Kurome Therapeutics. The remaining authors declare no competing financial interests.
Correspondence: Mohammad Azam, Cincinnati Children’s Hospital Medical Center, 240 Albert Sabin Way, Room S7.601, Cincinnati, OH 45229; e-mail: mohammad.azam@cchmc.org.

