

Key Points
This is the first report of long-term survival of a patient with FIP1L1-RARA–associated neoplasm treated with intensive multiagent therapy.
Integration of various genomic analyses may reveal unique genetic mechanisms of rare RARA fusions and its associated phenotypic features.
Abstract
FIP1L1-RARA–a ssociated neoplasm is a very rare and aggressive disease, with only 3 previously reported cases in the literature. Here, we describe a 9-month-old boy who presented with a FIP1L1-RARA fusion–associated myelodysplastic/myeloproliferative neoplasm-like overlap syndrome, with similarities and distinct features to both acute promyelocytic leukemia and juvenile myelomonocytic leukemia. Using a combined approach of chemotherapy, differentiating agents, and allogeneic hematopoietic stem cell transplant (allo-HCT), this patient remains in remission 20 months after allo-HCT. To our knowledge, this is only the second published pediatric case involving this condition and the only case with a favorable long-term outcome. Given the aggressive disease described in the previously published case report, as well as the successful treatment course described, the combinatorial use of chemotherapy, differentiation therapy, and allo-HCT for treatment of FIP1L1-RARA fusion–associated myeloid neoplasms should be considered.
Introduction
Oncogenic fusions of the retinoic acid receptor α (RARA) gene are the defining molecular features of acute promyelocytic leukemia (APL), with promyelocytic leukemia gene (PML) as the partner gene in 95% of the cases. Rarely, variant fusion partner genes are involved including PLZF, NPM1, NUMA, BCOR, OBFC2A, STAT5b, PRKAR1A, and FIP1L1.1,2 Incorporation of differentiation therapy with all-trans retinoic acid (ATRA) is standard for patients with APL based on robust response rates in both frontline and relapsed disease.2,3 Alternative fusion types demonstrate varying response rates to ATRA, with some fusion types being insensitive.4,5 More recently, synthetic retinoids with increased differentiating activity such as tamibarotene have been developed and have demonstrated potential clinical benefit.6
FIP1L1-RARA–associated leukemia is a rare alternative fusion type associated with aggressive disease. To date, 3 patients have been reported in the literature, 2 of whom died of disease.7-9 The first patient, a 20-month-old child with a diagnosis of juvenile myelomonocytic leukemia (JMML), died after multiple hematopoietic stem cell transplant (HCT) attempts 17 months after presentation. No mutations in PTPN11, K-Ras, or N-Ras were detected by Sanger sequencing of this child’s leukemia.7 The second reported case involved a 90-year-old woman diagnosed with APL who reportedly achieved remission after ATRA therapy, without further follow-up information.8 The third patient, a 77-year-old woman whose disease was also reported as APL, died 10 days after the start of therapy, possibly resulting from differentiation syndrome.9 Supplemental Table 1 provides patient characteristics of all reported cases, including the current case. The study was approved by the Institutional Review Board of the Memorial Sloan-Kettering Cancer Center.
Results
Case report
A previously healthy 9-month-old boy presented with persistent fevers and recurrent episodes of otitis media. Physical examination was significant for marked hepatosplenomegaly (spleen palpated below the umbilicus). A complete blood count demonstrated leukocytosis (white blood cells [WBCs] 22.8 K/μL), anemia (hemoglobin 9.3 g/dL), and mild thrombocytopenia (144 K/μL), with no evidence of bruising or coagulopathy (prothrombin time 14.1; international normalized ratio of 1.07; activated partial thromboplastin time 22.6). The peripheral blood cell differential revealed a predominance of abnormal myeloid precursor cells (64%), including promyelocyte-like and myelomonocytic forms that were difficult to classify but showed nonspecific esterase positivity, marked dysgranulopoiesis (>10%) including hypogranulation and abnormal nuclear segmentation, and monocytosis (14%; Figure 1A). Hemoglobin electrophoresis revealed borderline increased hemoglobin F (2.2%). Bone marrow examination revealed a normocellular for age marrow with myeloid hyperplasia and left-shifted maturation characterized by an abundance of hypergranular promyelocytic/myelomonocytic immature cells without evidence of Auer rods or other features associated with classical APL, and dysgranulopoiesis (>10%; Figure 1B,C). Dysplastic megakaryocytes (>10%) were also noted, whereas erythroid precursors were markedly decreased. Flow cytometry analysis showed an expanded population (90.5% of total WBCs) of immature myelomonocytic cells positive for CD64, CD14, bright CD123, with decreased expression of CD15 and HLA-DR, and a minute CD34+ population (0.084% of WBCs) with mild immunophenotypic atypia (Figure 1B-D; supplemental Figure 1). Conventional karyotype analysis demonstrated a t(4;17)(q12;q21) in 15 of 20 analyzed metaphase cells as the sole abnormality, which results in the FIP1L1-RARA fusion, confirmed by fluorescent in situ hybridization (FISH) analysis, and further characterized by reverse transcriptase polymerase chain reaction (PCR) and targeted RNA sequencing as an in-frame fusion between exon 13 of FIP1L1 and exon 3 of RARA (Figure 1E,F).

Peripheral blood and bone marrow evaluation of the patient’s leukemic cells at diagnosis. (A) Wright-Giemsa–stained peripheral blood smear (×100 magnification) at diagnosis, showing abnormal promyelocytes, immature myelomonocytic cells (arrow), and dysplastic neutrophils. (B) Hematoxylin and eosin–stained bone marrow biopsy (×20 magnification; inset ×4) at diagnosis. (C) Bone marrow aspirate smear (×100 magnification). Arrows indicate the dysplastic granulocytes. (D) Flow cytometry of diagnostic bone marrow aspirate. (E) Fusion schematic as identified by RNA sequencing. (F-G) Flow-sorted FISH: FIP1L1-RARA fusion was evaluated in granulocytic cells and CD34+ cells by triple color and RARA break-apart FISH. DBD, DNA-binding domain; LBD, ligand-binding domain; NLS, nuclear localization signal.
Peripheral blood and bone marrow evaluation of the patient’s leukemic cells at diagnosis. (A) Wright-Giemsa–stained peripheral blood smear (×100 magnification) at diagnosis, showing abnormal promyelocytes, immature myelomonocytic cells (arrow), and dysplastic neutrophils. (B) Hematoxylin and eosin–stained bone marrow biopsy (×20 magnification; inset ×4) at diagnosis. (C) Bone marrow aspirate smear (×100 magnification). Arrows indicate the dysplastic granulocytes. (D) Flow cytometry of diagnostic bone marrow aspirate. (E) Fusion schematic as identified by RNA sequencing. (F-G) Flow-sorted FISH: FIP1L1-RARA fusion was evaluated in granulocytic cells and CD34+ cells by triple color and RARA break-apart FISH. DBD, DNA-binding domain; LBD, ligand-binding domain; NLS, nuclear localization signal.
The patient received high-risk APL therapy based on the identification of the variant RARA fusion along with an elevated WBC count. He received induction therapy with ATRA and arsenic trioxide (ATO) combined with idarubicin. He achieved a morphologic remission after induction with minimal residual disease detectable by FISH. Following a cycle with differentiation therapy alone (ATRA and ATO), he achieved a cytogenetic remission. After a consolidation cycle with mitoxantrone and cytarabine, he was started on maintenance therapy with ATRA and ATO with a planned 6-month duration. During the last month of planned maintenance therapy, he presented with new-onset facial swelling and palpable skull-based masses. A complete blood count at that time revealed an increase in peripheral blood promyelocytes and thrombocytopenia. Magnetic resonance imaging of the cranium revealed extensive expansile multicompartmental disease with skull base, sinonasal, orbital, and mandible spread (Figure 2A). Bone marrow aspiration demonstrated replacement of the marrow with the previously diagnosed myeloid neoplasm with confirmation of FIP1L1-RARA fusion.
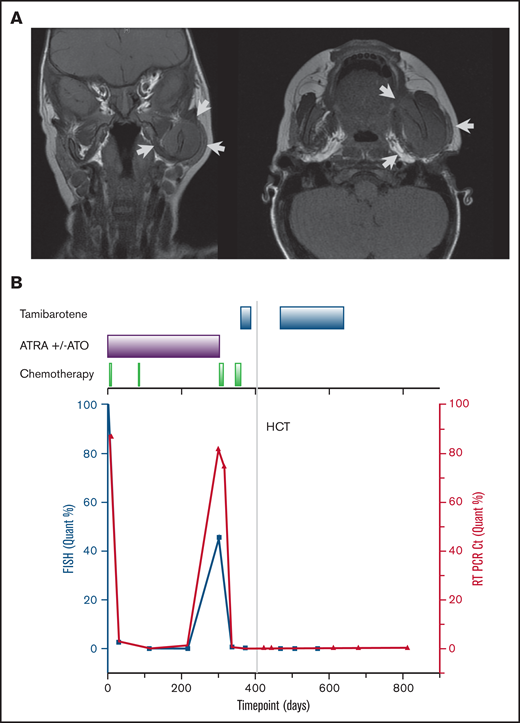
Imaging findings at the time of relapse and overall patient’s treatment course. (A) Magnetic resonance imaging of the skull at the time of relapse. Arrows indicate areas of disease. (B) Clinical treatment course (top axis) and disease burden as represented by FIP1L1-RARA fusion identified by FISH cells (left y-axis) and quantitative PCR (right y-axis).
Imaging findings at the time of relapse and overall patient’s treatment course. (A) Magnetic resonance imaging of the skull at the time of relapse. Arrows indicate areas of disease. (B) Clinical treatment course (top axis) and disease burden as represented by FIP1L1-RARA fusion identified by FISH cells (left y-axis) and quantitative PCR (right y-axis).
The patient was treated with the Capizzi II regimen (high-dose cytarabine) reinduction therapy, which resulted in marked improvement of the skull chloromas and morphologic marrow remission with residual disease identified by FISH. In an attempt to achieve minimal residual disease (MRD) negativity before proceeding to HCT, he received topotecan, vinorelbine, thiotepa, and clofarabine chemotherapy combined with the synthetic retinoid agent tamibarotene .10 Following count recovery, marrow assessment demonstrated MRD negativity by FISH. He proceeded to a CD34 selected peripheral blood stem cell transplant (HCT) from an unrelated HLA-identical donor in April 2019 following cytoreduction with rabbit ATG, clofarabine, melphalan, and thiotepa. He received an additional 3 months of maintenance therapy with tamibarotene started day +60 post-HCT. He remains in cytogenetic remission 20 months after HCT. A summary of his disease course is depicted in Figure 2B.
APL is characterized by the presence of a RARA translocation that leads to maturation arrest of the myeloid progenitor cells at the promyelocytic stage.11 In this case, a variant FIP1L1-RARA fusion was identified along with an abundance of promyelocytes with atypical morphology and immature myelomonocytic forms. Integration of various cytogenetic and genomic assays enabled us to better understand the mechanisms of this rare FIP1L1-RARA fusion and its associated leukemia. FISH analysis of flow-sorted cell populations confirmed this fusion in the abnormal myelomonocytic cells, and not in the minute and distinct CD34+ stem/progenitor cells, which showed only mild immunophenotypic atypia (Figure 1F,G). A final diagnosis of FIP1L1-RARA fusion–associated myeloid neoplasm was assigned, with a differential diagnosis of variant APL vs an overlap myelodysplastic/myeloproliferative neoplasm (MDS/MPN) within the spectrum of JMML. Next-generation sequencing-based mutational profiling (MSK-IMPACT Heme)12 targeting 400 genes altered in hematological malignancies was performed with a paired matched normal sample and revealed a somatic MAP2K2 p.R231L mutation (23.5% allele frequency).
The sensitivity of FIP1L1-RARA to ATRA has been demonstrated in prior in vitro studies.8 Similarly, leukemic cells collected from this patient at diagnosis demonstrated in vitro sensitivity to both ATRA and tamibarotene (supplemental Figure 2). Furthermore, there was clinical evidence of sensitivity as ATRA and ATO eradicated postinduction MRD.
MRD monitoring of PML-RARA using quantitative PCR (qPCR) has been well established for risk stratification and identification of early relapse in patients with APL.13,14 Because a clinical qPCR assay for the FIP1L1-RARA fusion was not available for this patient, FISH was used for MRD monitoring. During the treatment course, a research-based qPCR assay for the FIP1L1-RARA fusion was designed and performed on banked samples. As shown in Figure 2B, there was concordance between clinical FISH testing and qPCR results.
Discussion
Here, we describe an extensive workup and successful treatment of a child with FIP1L1-RARA fusion–associated MDS/MPN neoplasm overlap syndrome. The absence of the FIP1L1-RARA fusion in CD34+ stem/progenitor cells raise a diagnostic consideration of APL. However, several features were not consistent with typical APL, including: absence of laboratory or clinical signs of coagulopathy, very young age, splenomegaly, myelodysplastic features seen in the bone marrow histopathology, monocytic differentiation of the immature cells (bright CD11b expression, uniform CD14 expression), expression of intermediate to bright CD15, and a somatic mutation in a gene involved in mitogen-activated protein kinase signaling. This patient’s presentation as an infant with leukocytosis characterized by immature myeloid cells with monocytic differentiation, a borderline increased hemoglobin F, an absolute monocyte count of >1000/μL, as well as the marked splenomegaly, are suggestive of a diagnosis of JMML. Genomic profiling identified FIP1L1-RARA as the driver oncogene, without evidence of an established oncogenic RAS pathway mutation or monosomy 7. However, a somatic MAP2K2 missense mutation was detected, which is of uncertain significance. This variant has been identified as a germline alteration in at least 3 independent occurrences in patients with clinical RASopathies and Noonan syndrome,15 and based on computational predictions and evolutionary conservation analysis (SIFT, PolyPhen-2, Align-GVGD),16-19 it is predicted to impact the protein. MAP2K2 encodes the MEK2 kinase that is part of RAS pathway, which is commonly aberrantly activated in human cancers, including JMML. Mutations in MAP2K2 are infrequent in cancer (0.7% of predominantly solid tumors analyzed), found in a variety of tumor types, and spread across the length of the protein.20 Although there are no functional data on the exact missense mutation seen in our patient, a recent comparative study identified MAP2K2 p.R231H as a significant hot spot based on an in silico analysis, although there was no downstream ERK activation in vitro in transfected human embryonic kidney cells to suggest that this alteration is activating.20 It is unclear if the in vitro data can be extrapolated to signaling in hematopoietic cells in vivo and if a substitution by leucine rather than histidine is comparable in this regard. Thus, although the FIP1L1-RARA fusion is the oncogenic driver, the MAP2K2 mutation suggests that the biology of this patient’s malignancy may align in part with JMML, similar to the prior report of a pediatric patient with this fusion.7 Although this patient’s disease has clinical and biologic similarities to both APL and JMML, it has distinct features different from both disorders which necessitates its categorization as a unique myeloid neoplasm on the spectrum between these 2 entities that may be best categorized as MDS/MPN.
Unfortunately, our patient experienced relapsed disease while receiving maintenance therapy with ATRA and ATO. Large panel next-generation sequencing of the relapse sample did not reveal any RARA alterations that could explain resistance to therapy. Based on a recent phase 2 study demonstrating a 64% overall response rate of tamibarotene in patients with relapsed APL after treatment with ATRA and ATO,21 we opted to obtain tamibarotene through a single-patient expanded access mechanism for our patient. Because the drug was started immediately after topotecan, vinorelbine, thiotepa, and clofarabine chemotherapy without a marrow assessment in between, it is difficult to assess the degree of clinical benefit of tamibarotene. Similarly, we are unable to assess its benefit as post-HCT maintenance therapy. However, the combination of tamibarotene and chemotherapy led to a second MRD− complete remission before HCT, and our patient remains in remission 20 months after HCT.
In conclusion, this case adds to the scant published literature on this ultrarare disorder. Given the aggressiveness described in previously published case reports, as well as the successful treatment course described here, the combinatorial use of chemotherapy, differentiation therapy, and allogeneic HCT for treatment of FIP1L1-RARA fusion associated myeloid neoplasms should be considered.
Acknowledgment
This work was supported by the Scarlett Fund in part by the National Institutes of Health National Cancer Institute Cancer Center Support Grant (P30 CA008748).
Authorship
Contribution: O.M. and N.S. provided direct patient care, performed analyses, and wrote the paper; S.K. and S.E.P., provided direct patient care, diagnosis, and edited the paper; K.P.-D., M.K., J.T.G., S.M., G.I., E.S., M. Roshal, and Y.Z. performed analysis, diagnostic evaluation, and edited the paper; M. Richardson and W.A. performed experimental analysis; N.K., R.B., J.B., D.Y., D.L., A.L.K., M. Roshal, and Y.Z. helped with the design, performance of the experiments, and edited the paper; and N.B. and I.R.-S. obtained clinical information and helped write the paper.
Conflict-of-interest disclosure: R.B. has received a grant and travel credit from ArcherDx, honoraria for advisory board participation from Loxo Oncology, and speaking fees from Illumina. S.E.P. receives support for the conduct of sponsored trials through MSK from Jasper, Atara Biotherapeutics, and AlloVIr and is an inventor of intellectual property licensed to Atara by MSK with all of her rights assigned to MSK. W.A. has stock ownership in Notable Labs. The remaining authors declare no competing financial interests.
Correspondence: Neerav Shukla, Department of Pediatrics, Memorial Sloan Kettering Cancer Center, 1275 York Ave, New York, NY 10065; e-mail: shuklan@mskcc.org.

