

Key Points
Efanesoctocog alfa is a new class of FVIII replacement that breaks the von Willebrand factor–imposed FVIII half-life ceiling.
Once-weekly efanesoctocog alfa was well tolerated and provided high sustained FVIII activity with a mean half-life of 37 to 41 hours.

Abstract
Efanesoctocog alfa (rFVIIIFc-VWF-XTEN; BIVV001) is a new class of factor VIII (FVIII) replacement that breaks the von Willebrand factor–imposed FVIII half-life ceiling. In a phase 1/2a study, single-dose efanesoctocog alfa was well tolerated, and no safety concerns were identified. We evaluated the safety, tolerability, and pharmacokinetics of repeat-dose efanesoctocog alfa in a phase 1 study in previously treated adults (≥150 exposure days) with severe hemophilia A. Participants received 4 once weekly doses of efanesoctocog alfa (cohort 1, 50 IU/kg; cohort 2, 65 IU/kg). All enrolled participants (cohort 1, n = 10; cohort 2, n = 14) completed the study. Inhibitor development to FVIII was not detected. After the last dose of efanesoctocog alfa, geometric mean (range) FVIII activity half-life, area under the activity-time curve, and steady-state maximum concentration for cohort 1 and cohort 2 were 41.3 (34.2-50.1) and 37.3 (28.9-43.8) hours, 8290 (5810-10 300) and 11 200 (7040-15 800) hours × IU/dL, and 131 (96-191) and 171 (118-211) IU/dL, respectively. There was minimal accumulation after 4 doses. Mean FVIII activity for cohort 1 and cohort 2, respectively, was 46% and 69% on day 3 postdose and 10% and 12% on day 7 postdose. Overall, 4 once-weekly doses of efanesoctocog alfa were well tolerated, no safety concerns were identified, and no bleeds were reported during the treatment period. Once-weekly efanesoctocog alfa provided high sustained FVIII activity within the normal to near-normal range for 3 to 4 days postdose and may improve protection against bleeds in patients with hemophilia A. The trial is study 2018-001535-51 in the EU Clinical Trials Register.
Introduction
Primary prophylaxis is the standard of care in severe hemophilia A, aiming to reduce bleeding occurrence and prevent joint damage.1 Historically, the goal of prophylaxis has been the maintenance of factor VIII (FVIII) activity trough levels between 1% and 3%.2 For standard half-life (SHL) factor replacement products (eg, recombinant FVIII [rFVIII]), this target level usually requires injections 3 times per week or every other day, whereas injections are given every 3 to 5 days for extended half-life (EHL) FVIII products.3-8 Target FVIII levels during prophylaxis are being reevaluated as more data suggest that, for many patients, higher sustained factor levels may be required to improve protection from bleeds, preserve joint health, and advance closer to the goal of a functional cure and healthy equity.9-13 Achieving these higher sustained factor activity levels with once-weekly dosing is not possible with currently approved FVIII therapies. Although pegylation and Fc fusion techniques have increased the half-life of current EHL FVIII products, the von Willebrand factor (VWF) chaperone effect that noncovalently stabilizes and protects endogenous and exogenous FVIII from early degradation limits FVIII half-life extension to 1.5-fold to two-fold that of SHL products.1,14-16 Achieving high sustained FVIII activity with once-weekly dosing is ultimately dependent upon decoupling FVIII from endogenous VWF.17
Efanesoctocog alfa (BIVV001) is a novel fusion protein that is designed to decouple rFVIII from endogenous VWF in circulation. It is composed of a single rFVIII protein fused to dimeric Fc, the D′D3 domain of VWF (FVIII-binding domain), and 2 XTEN polypeptides (XTEN is a registered trademark of Amurix Pharmaceuticals, Inc).17,18 Appending the D′D3 domain of VWF to FVIII in efanesoctocog alfa prevents binding to endogenous VWF and removes the limit on half-life extension imposed by VWF-FVIII interaction.17 The Fc fusion and XTEN technologies provide further half-life extension.19,20 In a phase 1/2A study, single-dose efanesoctocog alfa provided high sustained factor activity levels with once-weekly dosing and was well tolerated; no safety concerns were identified.18 After an injection of 65 IU/kg efanesoctocog alfa, FVIII activity was within the normal range (ie, >50%) at day 4, and it was 17% at day 7. The mean elimination half-life (t1/2) and area under the FVIII-time curve (0 to infinity) for 65 IU/kg efanesoctocog alfa was threefold to fourfold longer and six- to sevenfold greater, respectively, than for rFVIII. Because efanesoctocog alfa is intended to be used as a once-weekly prophylactic therapy, the objective of the current study was to assess the safety, tolerability, and pharmacokinetics (PK) of repeat doses of efanesoctocog alfa in participants with severe hemophilia A.
Methods
Study population and study design
This was a phase 1 open-label single-site study to evaluate the safety, tolerability, and PK of 4 once-weekly doses of efanesoctocog alfa in adult males between 18 and 65 years of age with severe hemophilia A (<1 IU/dL [<1%] endogenous FVIII at screening) who had received ≥150 exposure days of prior FVIII treatment. Key exclusion criteria included a personal history of a positive FVIII inhibitor test or clinical signs of decreased response to FVIII administrations, a plasma VWF ristocetin cofactor level < 50 IU/dL, other known coagulation disorders in addition to hemophilia A, and a history of hypersensitivity or anaphylaxis associated with any FVIII product.
All participants provided informed consent prior to enrollment, and the study was conducted in accordance with international ethics guidelines, including the Declaration of Helsinki, and the International Council for Harmonization of Technical Requirements for Pharmaceuticals for Human Use, guidelines for Good Clinical Practice, and all applicable local laws, rules, and regulations. The local Institutional Review Board was located at the Specialized Hospital for Active Treatment of Hematological Diseases. The national Institutional Review Board was the Ethics Committee for Multicenter Clinical Trials (Sofia, Bulgaria).
A total of 24 participants were enrolled in the study: 10 participants in cohort 1 (50 IU/kg efanesoctocog alfa) and 14 participants in cohort 2 (65 IU/kg efanesoctocog alfa). The study consisted of a screening and washout period, a dosing period, and a safety observation period (Figure 1).

Efanesoctocog alfa repeat-dose study design. The study design was the same for both cohorts (50 or 65 IU/kg once weekly). Blood samples for PK trough measurements were obtained before dosing on days 8, 15, and 22.
Efanesoctocog alfa repeat-dose study design. The study design was the same for both cohorts (50 or 65 IU/kg once weekly). Blood samples for PK trough measurements were obtained before dosing on days 8, 15, and 22.
Participants were enrolled to receive a total of 4 once-weekly doses of efanesoctocog alfa (50 or 65 IU/kg) on days 1, 8, 15, and 22. Enrollment in the lower-dose cohort was completed first, followed by enrollment of a separate group of participants in the higher-dose cohort. The doses and dosing interval were primarily selected based on previous clinical study results18 and to achieve a steady-state efanesoctocog alfa PK profile with high sustained factor levels.
Blood samples for measuring FVIII activity and subsequent PK analysis were collected immediately before each injection of efanesoctocog alfa and at 0.5, 3, 24, 28, 72, 120, and 168 hours (predose at day 8) after the first dose and at 0.5, 3, 24, 48, 72, 120, 168, 240, and 336 hours after the last dose. FVIII activity was determined using an activated partial thromboplastin time–based 1-stage clotting assay (Dade Actin FSL Activated PTT Reagent; Siemens Healthcare), which was used for potency assignment. A 2-stage chromogenic substrate assay (BIOPHENTM Factor VIII:C assay; HYPHEN BioMed) was also used for FVIII activity measurement; both assays used a product-specific standard. Blood samples for FVIII inhibitor testing were collected at screening and on days 1, 8, 15, 22, 36, and 50. FVIII inhibitor testing was performed using the Nijmegen-modified Bethesda assay.
There was a 28-day safety observation period after the last dose of efanesoctocog alfa (day 22). Participants could resume treatment with their prestudy FVIII product halfway through this safety observation period after completing the day-36 visit (ie, 14 days after the final dose of efanesoctocog alfa). During the study, if applicable, the prestudy FVIII product was used in accordance with the local standard of care.
Study objectives and outcomes
The primary objective was to assess the safety and tolerability of 4 once-weekly IV doses of efanesoctocog alfa based on the occurrence of adverse events (AEs) and clinically significant abnormalities in laboratory tests, including the development of inhibitors to FVIII (neutralizing antibodies directed against FVIII, defined as a result ≥ 0.6 Bethesda unit per milliliter, identified and confirmed by a second test on an independent sample collected within 2 to 4 weeks of the first positive sample, with both tests performed by the central laboratory using the Nijmegen-modified Bethesda assay).
The secondary objective was to characterize the PK of efanesoctocog alfa after the 4 once-weekly IV doses for each cohort. Baseline-corrected FVIII activity was calculated by subtracting FVIII activity levels at baseline. FVIII activity was summarized by cohort, scheduled PK visit, and time point. FVIII activity-time profiles were constructed for each subject and the 2 cohorts, and PK analyses were performed using noncompartmental analysis (Phoenix® WinNonlin® 8.0). Calculated PK parameters included maximum FVIII activity, elimination half life interval (t1/2), total clearance at steady-state, accumulation index, area under the FVIII activity-time curve from hour 0 over the dosing interval (AUC0-τ), volume of distribution at steady-state, mean residence time, incremental recovery, and FVIII activity before the next administered dose. Accumulation index was defined as the ratio of AUC0-τ at steady-state/AUC0-τ after the first dose.
Prestudy treatment regimen, as well as medical and bleeding history, were captured as documented in medical records at screening. On-study occurrence of bleeding episodes was recorded in the electronic case report form.
Statistical analysis
Descriptive statistics were used to summarize demographic and clinical characteristics by dose cohort. No formal statistical hypothesis was tested. All safety analyses were performed using the safety analysis set, which included all participants who received ≥1 dose of efanesoctocog alfa. Safety and PK data were summarized by cohort using standard summary statistics for continuous and categorical data; PK parameters were summarized using the geometric mean and 95% confidence interval. Actual body weight was used to calculate dose. PK parameters are presented for day 1 (after a single dose) and day 22 (at steady-state). AEs were coded using the Medical Dictionary for Regulatory Activities System Organ Classes and Preferred Terms (version 21).
Results
Participant disposition and baseline characteristics
Twenty-four participants were enrolled in this study (10 in cohort 1 [50 IU/kg] and 14 in cohort 2 [65 IU/kg]); all enrolled participants completed the study (Table 1). Of the 24 participants, 23 received the protocol-specified efanesoctocog alfa dose and had adequate blood sample collection to allow for PK assessment (9 in cohort 1 and 14 in cohort 2). The mean age of participants was 35 years (range, 25–55) in cohort 1 and 41 years (range, 24-58) in cohort 2 (Table 1). The median number of bleeds within the 12 months prior to screening was 48 (range, 1-65) and 48 (range, 3-96) in cohorts 1 and 2, respectively (Table 1). Prior to study entry, most participants (21; 88%) were receiving on-demand FVIII treatment (Table 1). No participant had a family history of inhibitors.
Participant disposition, demographics, and baseline clinical characteristics
| Characteristic | Cohort 1 (50 IU/kg efanesoctocog alfa) | Cohort 2 (65 IU/kg efanesoctocog alfa) |
|---|---|---|
| Enrolled, n | 10 | 14 |
| Included in safety analysis* | 10 (100) | 14 (100) |
| Included in pharmacokinetic analysis† | 9 (90) | 14 (100) |
| Completed study‡ | 10 (100) | 14 (100) |
| Age, mean (range), y | 35 (25-55) | 41 (24-58) |
| Weight, mean (range), kg | 79.3 (51.0-113.0) | 95.3 (53.0-130.9) |
| White race | 10 (100) | 14 (100) |
| Ongoing or resolved medical/surgical history§ | ||
| Hepatic | 7 (70) | 13 (92.9) |
| Musculoskeletal | 9 (90) | 11 (78.6) |
| Cardiovascular | 1 (10) | 8 (57.1) |
| Gastrointestinal | 2 (20) | 2 (14.3) |
| Other | 0 (0) | 2 (14.3) |
| Time since hemophilia diagnosis, mean (SD), y | 34.1 (9.9) | 38.8 (9.7) |
| No. of bleeds in past 12 mo, median (range) | 48 (1-65) | 48 (3-96) |
| Prestudy FVIII regimen¶ | ||
| On demand | 8 (80) | 13 (92.9) |
| Prophylaxis | 2 (20) | 1 (7.1) |
| Characteristic | Cohort 1 (50 IU/kg efanesoctocog alfa) | Cohort 2 (65 IU/kg efanesoctocog alfa) |
|---|---|---|
| Enrolled, n | 10 | 14 |
| Included in safety analysis* | 10 (100) | 14 (100) |
| Included in pharmacokinetic analysis† | 9 (90) | 14 (100) |
| Completed study‡ | 10 (100) | 14 (100) |
| Age, mean (range), y | 35 (25-55) | 41 (24-58) |
| Weight, mean (range), kg | 79.3 (51.0-113.0) | 95.3 (53.0-130.9) |
| White race | 10 (100) | 14 (100) |
| Ongoing or resolved medical/surgical history§ | ||
| Hepatic | 7 (70) | 13 (92.9) |
| Musculoskeletal | 9 (90) | 11 (78.6) |
| Cardiovascular | 1 (10) | 8 (57.1) |
| Gastrointestinal | 2 (20) | 2 (14.3) |
| Other | 0 (0) | 2 (14.3) |
| Time since hemophilia diagnosis, mean (SD), y | 34.1 (9.9) | 38.8 (9.7) |
| No. of bleeds in past 12 mo, median (range) | 48 (1-65) | 48 (3-96) |
| Prestudy FVIII regimen¶ | ||
| On demand | 8 (80) | 13 (92.9) |
| Prophylaxis | 2 (20) | 1 (7.1) |
Unless otherwise noted, data are n (%).
SD, standard deviation.
The safety analysis included participants who received ≥1 dose of efanesoctocog.
The PK analysis included participants who had adequate blood samples for PK assessments, as determined by the sponsor.
Defined as participants who did not discontinue early.
Includes conditions present in >10% of participants in either group.
The most recent regimen that the participant was receiving prior to the study.
Safety
All 24 participants received ≥1 dose of efanesoctocog alfa and were included in the safety analysis set. During the treatment period, 12 (50%) participants (8/10 participants [80%] in cohort 1 and 4/14 participants [29%] in cohort 2) reported ≥1 treatment-emergent AE (TEAE). A total of 25 TEAEs (19 in cohort 1 and 6 in cohort 2) were reported, of which 23 were assessed by the investigator as mild in severity and 2 were assessed as moderate. There were no reports of study drug–related TEAEs, TEAEs leading to discontinuation, or treatment-emergent serious AEs. The most common (≥2 participants overall) TEAEs reported were rhinitis (n = 4; 17%), arthralgia (n = 2; 8%), upper respiratory tract infection (n = 2; 8%), and headache (n = 2; 8%). No inhibitor development to FVIII was detected up to 28 days following the final dose of efanesoctocog alfa. In addition, there were no reports of hypersensitivity, anaphylaxis, or vascular thrombotic events.
PK
PK data were available for 9 participants in cohort 1 and all 14 participants in cohort 2. One participant in cohort 1 received a partial dose during the first infusion and received full doses for the next 3 infusions (supplemental Material). This participant was excluded from the PK analysis set. On day 22, the mean FVIII activity for cohorts 1 and 2 was 46% and 69% at 72 hours, 22% and 27% at 120 hours, and 10% and 12% at 168 hours, respectively, with mean FVIII activity in the normal to near-normal range (>40%) for 3 to 4 days postdose (Figure 2).
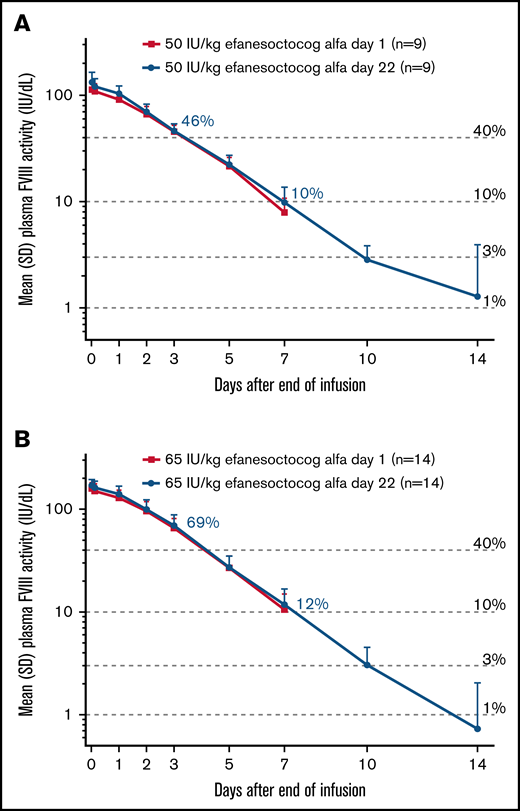
Baseline-corrected factorVIII activity over time. Data are mean ± standard deviation (SD) for cohort 1 (A) and cohort 2 (B) and are based on the 1-stage activated partial thromboplastin time clotting assay. †Values are for factor VIII activity levels after the day-22 dose.
Baseline-corrected factorVIII activity over time. Data are mean ± standard deviation (SD) for cohort 1 (A) and cohort 2 (B) and are based on the 1-stage activated partial thromboplastin time clotting assay. †Values are for factor VIII activity levels after the day-22 dose.
Baseline-corrected PK data for days 1 and 22, based on the activated partial thromboplastin time 1-stage clotting assay, are summarized in Table 2. On day 22 (steady-state), geometric mean t1/2 was 41.3 and 37.3 hours in cohorts 1 and 2, respectively. Geometric mean maximum FVIII activity (Cmax) and AUC0-τ at steady-state were 131 and 171 IU/dL and 8290 and 11 200 h × IU/dL for cohorts 1 and 2, respectively, and indicate that Cmax and AUC0-τ are dose proportional at these doses. Geometric mean clearance at-steady state (CLss) and mean residence time extrapolated to infinity (MRTinf for 50 IU/kg efanesoctocog alfa were 0.60 mL/h per kilogram and 62.3 h, respectively, and both were consistent across cohorts. Incremental recovery on day 22 was 2.43 and 2.45 IU/dL per IU/kg in cohorts 1 and 2, respectively. Overlapping day-1 and day-22 mean FVIII activity profiles were consistent, with geometric mean accumulation indices of 1.05 and 1.07 (Table 2; Figure 2). Similar results were observed when PK parameters were derived from the 2-stage chromogenic assay (supplemental Table).
PK parameters
| Cohort 1 (50 IU/kg efanesoctocog alfa [n = 9]) | Cohort 2 (65 IU/kg efanesoctocog alfa [n = 14]) | |||
|---|---|---|---|---|
| Day 1* | Day 22 | Day 1* | Day 22 | |
| t1/2, h | — | 41.3 (37.0-46.1) | — | 37.3 (34.6-40.2) |
| Cmax, IU/dL† | 113 (98-130) | 131 (110-157) | 158 (144-174) | 171 (157-186) |
| AUC0-τ, h × IU/dL | 7650 (6750-8670) | 8290 (7260-9460) | 10 500 (9 230-12 000) | 11 200 (9 800-12 800) |
| CLss, mL/h/kg | — | 0.60 (0.53-0.69) | — | 0.58 (0.51-0.66) |
| IR, IU/dL per IU/kg | 2.26 (1.95-2.61) | 2.43 (2.03-2.92) | 2.43 (2.21-2.68) | 2.45 (2.26-2.66) |
| MRTinf, h | — | 62.3 (56.2-69.1) | — | 58.9 (55.7-62.2) |
| Accumulation index‡ | — | 1.07 (1.05-1.09) | — | 1.05 (1.04-1.06) |
| Cohort 1 (50 IU/kg efanesoctocog alfa [n = 9]) | Cohort 2 (65 IU/kg efanesoctocog alfa [n = 14]) | |||
|---|---|---|---|---|
| Day 1* | Day 22 | Day 1* | Day 22 | |
| t1/2, h | — | 41.3 (37.0-46.1) | — | 37.3 (34.6-40.2) |
| Cmax, IU/dL† | 113 (98-130) | 131 (110-157) | 158 (144-174) | 171 (157-186) |
| AUC0-τ, h × IU/dL | 7650 (6750-8670) | 8290 (7260-9460) | 10 500 (9 230-12 000) | 11 200 (9 800-12 800) |
| CLss, mL/h/kg | — | 0.60 (0.53-0.69) | — | 0.58 (0.51-0.66) |
| IR, IU/dL per IU/kg | 2.26 (1.95-2.61) | 2.43 (2.03-2.92) | 2.43 (2.21-2.68) | 2.45 (2.26-2.66) |
| MRTinf, h | — | 62.3 (56.2-69.1) | — | 58.9 (55.7-62.2) |
| Accumulation index‡ | — | 1.07 (1.05-1.09) | — | 1.05 (1.04-1.06) |
CLss, clearance at steady state; Cmax, maximum FVIII activity; IR, incremental recovery; MRT, mean residence time.
All data are geometric mean (95% confidence interval) and are based on the 1-stage activated partial thromboplastin time clotting assay.
Some parameters were not calculated for the first dose administered because they were more accurately calculated after the final dose when blood sampling could continue for 14 days postdose.
Day 22 value is Cmax at steady-state.
Accumulation index was defined as the ratio of AUC0-τ at steady-state/AUC0-τ after the first dose.
Bleeding events
No bleeds were reported during the 4-week treatment period with efanesoctocog alfa (defined as days 1 through 28 [7 days after the last dose of efanesoctocog alfa]). Nine participants in cohort 1 and 13 participants in cohort 2 experienced ≥1 bleeding episode on study; all bleeding episodes occurred after enrollment but before the first dose of efanesoctocog alfa or >1 week after the last dose of efanesoctocog alfa.
Discussion
FVIII replacement remains the most widely used therapy for severe hemophilia A. One of its advantages is that it can be used as a single therapy across many clinical scenarios (eg, treatment of bleeds, perioperative management, and prophylaxis).21 For prophylaxis, a key goal is to maintain joint health by preventing intra-articular bleeds. However, there is increasing evidence that target FVIII trough levels of 1% to 3% are insufficient to prevent the occurrence of all bleeds and progressive joint damage.9 For example, in the PROPEL study, more patients with severe hemophilia A did not have any bleeds if they achieved target trough FVIII levels of 8% to 12% compared with those reaching 1% to 3% during prophylaxis.10 Also, Warren et al22 reported that, despite starting prophylaxis at an early age, severe osteochondral damage developed in up to 35% of patients when assessed as adolescents. Accordingly, target FVIII levels for prophylaxis are being reevaluated to achieve a lower risk for all bleeds, optimal long-term joint protection, and improved quality of life for people living with hemophilia.11-13
Efanesoctocog alfa is a new class of FVIII replacement therapy that is designed to be independent of endogenous VWF and break the VWF-imposed FVIII t1/2 ceiling.17 In the current study, efanesoctocog alfa achieved high sustained FVIII activity with an elimination t1/2 that was substantially longer than that associated with current SHL and EHL FVIII products.1 Weekly treatment with efanesoctocog alfa provided normal to near-normal FVIII levels for up to 3 to 4 days following administration. The overlapping profiles of mean baseline–corrected FVIII activity over time for days 1 and 22 indicate minimal accumulation for the 50- and 65-IU/kg dosing groups. The results of this repeat-dose study are consistent with those observed for the single-dose study of efanesoctocog alfa.18
Four once-weekly doses of efanesoctocog alfa were well tolerated, and no safety concerns were identified. No inhibitor development to FVIII was detected, and there were no reports of hypersensitivity, anaphylaxis, or vascular thrombotic events. Although this study was not designed to assess efficacy, it is noteworthy that no bleeds were reported during the 4-week treatment period with efanesoctocog alfa. All bleeding episodes during the study occurred before dosing of efanesoctocog alfa or >1 week after the last dose.
High sustained FVIII activity is expected to provide high bleed protection.11,12 den Uijl et al demonstrated very low joint bleed rates in mild hemophilia when endogenous FVIII activity levels were ≥10% and, through modeling, predicted no joint bleeds when levels were ≥15%.11 Regression modeling by Soucie et al revealed that an endogenous FVIII activity level of 15% predicted only 1.4 joint bleeds per year for patients with nonsevere hemophilia A who were 25 to 44 years of age.12 These studies in nonsevere hemophilia evaluated bleed risk for people with relatively constant low endogenous factor VIII levels. In comparison, the administration of FVIII in severe hemophilia A aiming at achieving similar FVIII levels at the end of the dosing interval may provide better protection against bleeds as a result of sustained high factor levels in the normal to near-normal range during the first part of the dosing interval.
Overall, 4 once-weekly doses of efanesoctocog alfa were well tolerated and provided high sustained FVIII activity levels with minimal accumulation. These results suggest that efanesoctocog alfa may offer extended bleed protection with less frequent dosing for individuals with severe hemophilia A and support continued development of efanesoctocog alfa in ongoing phase 3 studies (NCT04161495 and NCT04644575).23
Acknowledgments
The authors thank Stella Lin for manuscript review and Dan Rubin and Joachim Fruebis for contributions to study design.
This work was supported by Sanofi and Sobi. Sanofi and Sobi reviewed and provided feedback on the manuscript. Editorial assistance was provided by Francis John Golder (Fishawack Communications Ltd., part of Fishawack Health) and was funded by Sanofi.
Authorship
Contribution: S.P. and S.K. analyzed the safety and PK data, respectively, and K.R. performed statistical analyses and all authors had access to primary clinical trial data, designed the study and/or acquired, analyzed, or interpreted data, and wrote/reviewed, had full editorial control of, and approved the final version of the manuscript.
Conflict-of-interest disclosure: A.W., S.K., S.P., and C.B., are employees of, and hold equity interest in, Sanofi. K.R. is an employee of Alexion, AstraZeneca Rare Disease. T.L. declares no competing financial interests.
Current affiliation for Kara Rice: Alexion, AstraZeneca Rare Disease, Boston, MA, USA.
Correspondence: Annemieke Willemze, Sanofi, Paasheuvelweg 25, 1105 BP Amsterdam, The Netherlands; e-mail: annemieke.willemze@sanofi.com.

