

Key Points
Metabolic rewiring promotes ATO resistance in APL, independent of PML mutation status.
Inhibition of mitochondrial respiration combined with ATO is a potential therapeutic option for relapsed APL and non-M3 AML.

Abstract
Acquired genetic mutations can confer resistance to arsenic trioxide (ATO) in the treatment of acute promyelocytic leukemia (APL). However, such resistance-conferring mutations are rare and do not explain most disease recurrence seen in the clinic. We have generated stable ATO-resistant promyelocytic cell lines that are less sensitive to all-trans retinoic acid (ATRA) and the combination of ATO and ATRA compared with the sensitive cell line. Characterization of these resistant cell lines that were generated in-house showed significant differences in immunophenotype, drug transporter expression, anti-apoptotic protein dependence, and promyelocytic leukemia-retinoic acid receptor alpha (PML-RARA) mutation. Gene expression profiling revealed prominent dysregulation of the cellular metabolic pathways in these ATO-resistant APL cell lines. Glycolytic inhibition by 2-deoxyglucose (2-DG) was sufficient and comparable to the standard of care (ATO) in targeting the sensitive APL cell line. 2-DG was also effective in the in vivo transplantable APL mouse model; however, it did not affect the ATO-resistant cell lines. In contrast, the resistant cell lines were significantly affected by compounds targeting mitochondrial respiration when combined with ATO, irrespective of the ATO resistance-conferring genetic mutations or the pattern of their anti-apoptotic protein dependency. Our data demonstrate that combining mitocans with ATO can overcome ATO resistance. We also show that this combination has potential for treating non-M3 acute myeloid leukemia (AML) and relapsed APL. The translation of this approach in the clinic needs to be explored further.
Introduction
Acute promyelocytic leukemia (APL) is a subtype of acute myeloid leukemia (AML) characterized by the presence of reciprocal translocation between the promyelocytic leukemia (PML) gene on chromosome 15 and the retinoic acid receptor alpha (RARA) gene on chromosome 17 [t(15;17)], resulting in the production of a chimeric and novel PML-RARΑ fusion oncoprotein that leads to blocking the differentiation of promyelocytes to mature granulocytes.1 Therapy with the combination of arsenic trioxide (ATO) and all-trans retinoic acid (ATRA)2-4 for managing APL has significantly improved survival rates when compared with treatment that uses ATO as a single agent or the combination of ATRA with chemotherapy.5-7
We recently reported on the mutational spectrum of patients with newly diagnosed or relapsed APL and demonstrated the importance of additional genetic events (FLT3, KRAS, NRAS, ARID1B, p53, and WT1) during disease recurrence. However, it was also noted that mutations resulting in primary or secondary resistance to ATO are extremely rare and could not explain the majority of disease relapses.8 Other mechanisms such as drug resistance mediated by the bone marrow microenvironment, upregulation of anti-apoptotic factors, modulation of cellular energy metabolism, and oxidative stress could potentially contribute to therapy resistance.
Existing literature on ATO resistance in APL has focused on the presence or acquisition of PML B2 domain mutations, with evidence supporting the presence of genetic mutations in the PML B2 domain (C212-S220; A216V) conferring resistance to ATO in APL. These mutations alter or inhibit ATO binding to the B2 domain of the PML component of the PML-RARA oncoprotein.9-13 However, such acquired somatic mutations are rarely seen in the clinic and cannot explain the relapses that occur in patients treated with ATO-based regimens. In addition, in APL, unlike the other subtypes of AML, there is little evidence to suggest the existence of a leukemic stem cell population that would explain disease recurrence.14
Recent observations and studies report the novel mechanism of action of ATO such as promotion of nonclassical apoptosis (ETosis: extracellular DNA traps) in a dose-dependent manner15 and inhibition of glycolysis. ATO directly binds to the Cys256 and Cys704 residues in hexokinase 2 (HK2) and pyruvate kinase (PKM2), which reduces the enzymatic activity of these proteins and acts as a glycolytic inhibitor.16 It has also been demonstrated that this glycolytic inhibition is an important mechanism by which ATO promotes apoptosis in cancer cells, and overexpression of HK2 significantly rescued the cells from ATO-induced apoptosis.16-18 These observations further illustrate that the mechanisms of action of ATO are complex and multifactorial, suggesting that mechanisms of resistance are also likely to be varied. To further interrogate the mechanisms of ATO resistance, we generated and characterized a stable ATO-resistant cell line with the objective of finding potentially druggable targets that could be used to overcome ATO resistance in APL.
Methods
Cell lines and chemicals
The human APL cell line NB4 was a kind gift from Harry Iland, MD, Royal Prince Alfred Hospital (Sydney, Australia) with permission from Michel Lanotte, MD. In addition to the in-house–generated ATO-resistant cell lines, we also used an ATRA-resistant APL cell line, UF1 (a kind gift from Christine Chomienne, MD, PhD, Hôpital Saint Louis, Paris, France). The cell lines were free from mycoplasma contamination (Universal Mycoplasma Detection Kit, American Type Culture Collection [ATCC], Manassas, VA). Primary cells were obtained after receiving written and informed consent from the patients (Institutional Review Board No: 5884). ATO was a gift from INTAS Pharmaceuticals (Ahmedabad, India). 2-NBDG (a fluorescent analog of D-glucose), JC-1, 2-deoxyglucose (2-DG), and carbonyl cyanide 4-(trifluoromethoxy) phenylhydrazone (FCCP) were purchased from Sigma-Aldrich (St. Louis, MO).
Exome sequencing
Genomic DNA was isolated from the naïve NB4 cells and the ATO-resistant NB4 subclones NB4EV-AsR1 and UF1 by using a Gentra Puregene Blood Kit (QIAGEN, Hilden, Germany) and was stored at 4°C. Library preparation and sequencing were performed at the Genomics Facility of Genotypic Technology (Bengaluru, India) after ion targetseq exome enrichment by the Ion Proton System. Sequencing was performed on the Ion Proton sequencer. Mutations were confirmed by Sanger sequencing.
Chromatin-immunoprecipitation sequencing (ChIP-seq)
H3K27ace pulldown was performed using simpleChIP Enzymatic Chromatin IP Kit per the manufacturer’s protocol (Cell Signaling Technology, Danvers, MA, USA). ChIP-seq data were generated from 2 independent experimental replicates. Total genomic DNA (input) derived from formaldehyde cross-linked samples was used as control during peak calling. Raw sequence reads that passed quality control were aligned to the human reference genome (available from the University of California-Santa Cruz genome browser; http://genome.ucsc.edu/). Peak calling on all ChIP-seq data were performed using MACS v2.1.
Gene expression array and analysis
A global gene expression array for differential gene expression in naïve NB4 cells and the ATO-resistant NB4 primary resistant clone was performed. NB4 naïve, NB4EV-AsR1, and UF1 cells (2 × 107) were harvested and stored in RNA later solution (Qiagen, Hilden, Germany). The extracted labeled RNAs were hybridized to Agilent Human Whole Genome 8 × 60K Gene Expression Array (AMADID: 039494), and Image analysis was performed using Agilent Feature Extraction software Version 10.5.1.1 to obtain the raw data. Normalization and statistical analysis of the microarray data were performed by using GeneSpring GX (Agilent Technologies, Santa Clara, CA, USA). Differentially regulated genes were clustered by using hierarchical clustering to identify significant gene expression patterns. Genes were classified on the basis of functions and pathways using the biological interpretation tool Biointerpreter (Genotypic Technology, Bangalore, Karnataka, India).
Seahorse extracellular flux analysis
Extracellular Flux Assay Kit XF24 (Agilent Technologies, Santa Clara, CA, USA) was used to measure oxygen consumption rate and glycolytic flux. Briefly, 3 replicate wells of 5 × 104 cells per well were seeded in a retronectin-coated (Takara Bio, Shiga, Japan) 24-well XF24 plate. At 30 minutes before analysis, the medium was replaced with Seahorse XF media (Agilent Technologies, Santa Clara, CA, USA), and the plate was incubated at 37°C. Analyses were performed at basal conditions and after injection of glucose, oligomycin, and 2-DG for glycolytic function.
Mouse model and drug treatments
FVB/N mice were obtained from The Jackson Laboratory (Bar Harbor, ME). Mice at 6 to 8 weeks of age were used in all the experiments. The animal study design and euthanasia protocols were approved by the Institutional Animal Ethics Committee (IAEC approval number 04/2019). APL cells from the spleen of MRP8-PML-RAR transgenic mice (FVB/N) were harvested and cryopreserved (a gift from Scott Kogan, MD [University of California-San Francisco). APL cells (106 cells per mouse) were injected IV via the tail vein into genetically compatible FVB/N recipients without conditioning. After the leukemic cell engraftment period (day 8), ATO (10 mg/kg) and 2-DG (750 mg/kg) were intraperitoneally injected for 15 days.
Intracellular BH3 (iBH3) profiling
First, 106 cells per mL were suspended in BH3 profiling buffer, digitonin permeabilized, and exposed to pro-apoptotic peptides at a concentration of 20 µM (Bim, BAD, HRK, and MS-1; GenScript, Piscataway, NJ, USA) for 90 minutes. We then proceeded according to the standardized protocol from the Anthony Letai laboratory (http://letailab.dana-farber.org/bh3-profiling.html) to measure intracellular retention of cytochrome c. After an overnight incubation with anti-cytochrome-c (6H2.B4) fluorescein isothiocyanate antibody (FITC) (BioLegend, San Diego, CA, USA), the cells were acquired on a flow cytometer (Beckman Coulter Navios, Brea, CA, USA). Loss of cytochrome c is proportional to the priming status of the mitochondria and its anti-apoptotic dependency by their peptide specificity.
Statistical analysis
All statistical analyses were carried out using GraphPad Prism 7.0 (GraphPad Software, La Jolla, CA, USA). All data points are represented as mean ± standard error of the mean. Two-tailed Student t test was used to compare mean values between 2 groups. One-way analysis of variance was used for experiments in which multiple groups were compared with control. P values < .05 were considered statistically significant. For further details and other methods, see supplemental Methods.
Results
Generation of ATO-resistant cell lines
ATO-resistant cells were generated by exposing the naïve NB4 cell line to low concentrations of ATO (50 nM) for about 3 months. The concentration of ATO was gradually increased to 1 µM over 1 year. The cells that survived and proliferated were termed “ATO-tolerant persister cells.” These ATO-tolerant persister cells were then subjected serially to limiting dilutions and single-cell colony-forming unit formation on methylcellulose to isolate monoclonal-resistant populations. We isolated 3 different clones, expanded them, and named them NB4EV-AsR1, NB4EV-AsR2, and NB4EV-AsR3 based on the published norms of NB4-resistant cell line nomenclature.19 The 50% inhibitory concentration (IC50) for ATO in these cell lines was 3.25 μM, 3.4 μM, and 2.88 μM for NB4EV-AsR1, NB4EV-AsR2, and NB4EV-AsR3, respectively, in contrast to naïve NB4 which was 0.9 µM (Figure 1A). The viability of the ATO-resistant cell lines generated in-house was not significantly affected by exposure to 2 μM ATO compared with the viability of the sensitive NB4 cell line (Figure 1B). The ATO-resistant cell lines generated in-house were also significantly less sensitive to the differentiation-inducing agent ATRA, similar to UF1, a known ATRA-resistant cell line. Even the combination of ATO (0.5 μM) and ATRA (1 μM) did not induce significant differentiation or cell death in the ATO-resistant cell lines generated in-house when compared with naive NB4 cells (Figure 1C). After exposure to ATRA (1 μM), the induction of downstream targets of the RAR-like TGM2, RARβ, and RARα transcripts was also found to be significantly less in the ATO-resistant cell lines generated in-house compared with NB4 naïve cells (supplemental Figure 1).
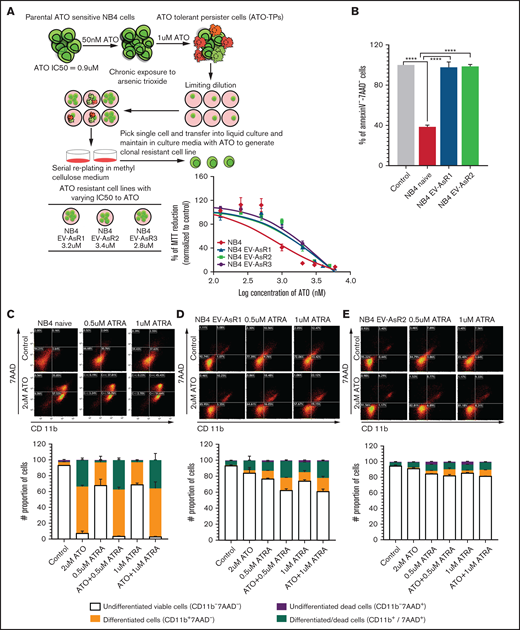
Generation of ATO-resistant cell lines. (A) The NB4 naïve parental cell line was exposed to 50 nM ATO for 3 months, and the concentration was gradually increased to 1 µM ATO over a period of 1 year until the cell population was sustained and proliferated. Limiting dilutions were used and colony-forming unit assays were performed to generate monoclones of the resistant cell lines. (B) The bar graph represents the percentage of viable cells after 48 hours of 2 µM ATO. (C) Representative dot plots and stacked bars (that summarize the dot plot results) for NB4 and resistant cell lines were treated with 0.5 µM and 1 μM of ATRA for 72 hours as single agents and in combination with 2 μM ATO. The percentage of differentiation was measured by the surface expression of CD11b, and dead cells were measured by 7-aminoactinomycin D (7-AAD). Graphs and statistical parameters were generated from 3 independent experiments. ****P ≤ .0001.
Generation of ATO-resistant cell lines. (A) The NB4 naïve parental cell line was exposed to 50 nM ATO for 3 months, and the concentration was gradually increased to 1 µM ATO over a period of 1 year until the cell population was sustained and proliferated. Limiting dilutions were used and colony-forming unit assays were performed to generate monoclones of the resistant cell lines. (B) The bar graph represents the percentage of viable cells after 48 hours of 2 µM ATO. (C) Representative dot plots and stacked bars (that summarize the dot plot results) for NB4 and resistant cell lines were treated with 0.5 µM and 1 μM of ATRA for 72 hours as single agents and in combination with 2 μM ATO. The percentage of differentiation was measured by the surface expression of CD11b, and dead cells were measured by 7-aminoactinomycin D (7-AAD). Graphs and statistical parameters were generated from 3 independent experiments. ****P ≤ .0001.
The doubling time of parental cell line NB4 was 28 hours, whereas for NB4EV-AsR1 and NB4EV-AsR2 clones, it was 46 and 48 hours, respectively. Long-term withdrawal of ATO from the culture system for 3 months did not result in the re-acquisition of ATO sensitivity (supplemental Figure 2). Currently, the resistant cell lines have a stable resistant phenotype when grown with or without ATO. We have observed that the ATRA-resistant UF1 cell line is also cross-resistant to ATO with an IC50 of 4.9 μM, an observation that has not been previously reported (supplemental Figure 3).
ATO-resistant cell lines exhibit distinct cell surface markers and transporters
The ATO-resistant cell lines were abnormal promyelocytes in which the cell surface expression of the typical myeloid markers CD13 and CD33 was significantly reduced compared with that in the parental naïve cell line (Figure 2A; supplemental Figure 4). The ATO efflux transporters such as AQP9, MRP4, ABCB6, and ABCA7 were significantly upregulated in the ATO-resistant cell lines, which mirrored the reduced concentration of intracellular ATO (Figure 2B-C).
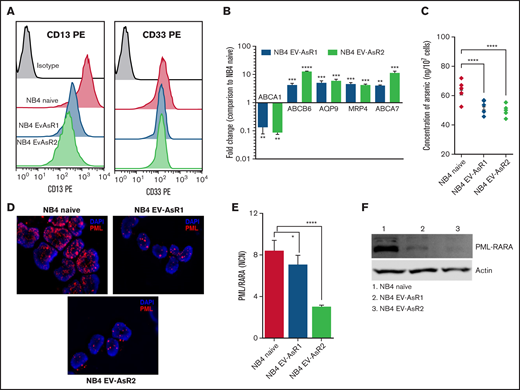
Heterogeneity in the cell surface marker and transporters expression of the ATO-resistant cell line compared with parental cell line. (A) CD13 and CD33 surface expression of ATO-resistant cell lines compared with the NB4 naïve cell line. (B) Relative messenger RNA (mRNA) levels of the ATO influx gene (ABCA1) and efflux transporters genes (ABCA7, AQP9, MRP4, and ABCB6) in the in-house–generated ATO-resistant cell lines compared with the NB4 naïve cell line which is normalized to 1. (C) Intracellular ATO levels in NB4 naïve and ATO-resistant cell lines after 24 hours of treatment with 0.5 µM ATO. (D) Fluorescent microscopic images of PML (red) in NB4 naïve and ATO-resistant cell lines that show nuclear body formation and micro speckled pattern (oil immersion lens; original magnification ×63). (E) Normalized copy number (NCN) of PML-RARA transcripts in the NB4 naïve and in-house–generated ATO-resistant cell lines. (F) Immunoblots of the PML-RARA fusion protein levels in NB4 naïve and ATO-resistant cell lines. All error bars represent the mean ± standard error of the mean (SEM) of 3 independent experiments. *P ≤ .05; **P ≤ .01; ***P ≤ .001; ***P ≤ .0001. DAPI, 4′,6-diamidino-2-phenylindole; PE, phycoerythrin.
Heterogeneity in the cell surface marker and transporters expression of the ATO-resistant cell line compared with parental cell line. (A) CD13 and CD33 surface expression of ATO-resistant cell lines compared with the NB4 naïve cell line. (B) Relative messenger RNA (mRNA) levels of the ATO influx gene (ABCA1) and efflux transporters genes (ABCA7, AQP9, MRP4, and ABCB6) in the in-house–generated ATO-resistant cell lines compared with the NB4 naïve cell line which is normalized to 1. (C) Intracellular ATO levels in NB4 naïve and ATO-resistant cell lines after 24 hours of treatment with 0.5 µM ATO. (D) Fluorescent microscopic images of PML (red) in NB4 naïve and ATO-resistant cell lines that show nuclear body formation and micro speckled pattern (oil immersion lens; original magnification ×63). (E) Normalized copy number (NCN) of PML-RARA transcripts in the NB4 naïve and in-house–generated ATO-resistant cell lines. (F) Immunoblots of the PML-RARA fusion protein levels in NB4 naïve and ATO-resistant cell lines. All error bars represent the mean ± standard error of the mean (SEM) of 3 independent experiments. *P ≤ .05; **P ≤ .01; ***P ≤ .001; ***P ≤ .0001. DAPI, 4′,6-diamidino-2-phenylindole; PE, phycoerythrin.
Because ATO induces the degradation of PML-RARA protein, we examined the subcellular localization, transcript, and protein levels of PML/PML-RARA in the resistant cells compared with that in the sensitive NB4 cell line. In the immunofluorescence assay, we observed that the PML localized in the nucleus in a typical APL-specific micro-speckled pattern in the NB4 cells, whereas in the resistant cell lines, we observed a similar micro-speckled pattern but a decrease in the amount of nuclear PML (Figure 2D). There was also significant reduction in the levels of PML-RARA at both the transcript and protein levels in the resistant cell lines (Figure 2E-F).
In-house–generated ATO-resistant cell lines harbor additional cytogenetic and molecular aberrations
Cytogenetic analysis of both cell lines showed triploidy as well as t(15;17). However, NB4EV-AsR1 showed additional cytogenetic abnormalities such as del5q, gain of chromosome 4, and loss of chromosome 22. The loss of the X chromosome as well as the absence of the derivative chromosome 21 and the addition 16q, which was seen in NB4 naïve cells, was not observed in NB4EV-AsR1 (supplemental Table 1).
Because there are reports implicating the emergence of somatic PML domain mutations in APL cells that confer drug resistance against ATO, we performed whole-exome sequencing on our in-house–generated ATO-resistant NB4EV-AsR1 cell line (as a representative of other ATO-resistant clones) compared with the NB4 naïve parental cell line and also on the UF1 cell line that was found to be resistant to ATO.
Whole-exome sequencing revealed that compared with the NB4 naïve cell line, a significant number of genes were mutated in the NB4EV-AsR1 and UF1 cell lines. We considered the mutation frequency and observed that compared with the NB4 naïve cells, the majority of the mutated genes in NB4EV-AsR1 belong to cell surface proteins, especially mucins (MUC6, MUC5B, MUC4, MUC3A, MUC16), serine protease genes (PRSS1, PRSS3, PRSS3P2), and the ATO resistance-conferring PML B2 domain mutation (A216V). In contrast, the UF1 cell line showed a higher frequency of mutations involving MUC16, ITGB4, PRSS1, CUL7, CDH23, LTBP3, OBSCN, and STAB1 genes (supplemental Figure 5) and did not have a PML B2 domain mutation. Further comparison of mutated genes in the resistant cell lines with the commonly observed mutations in the AML data set from The Cancer Genome Atlas (TCGA) revealed that in NB4EV-AsR1 (with the exception of the PML B2 domain mutation), we did not observe any additional novel mutations over and above those described in the AML TCGA gene set. However, in UF1, there were additional novel mutations found in DNMT3A, TP53, RUNX1, IDH2, SMC3, ARID1B, ARID1A, and PML genes in addition to the known mutations in the AML TCGA gene set (Figure 3A; supplemental File 1).
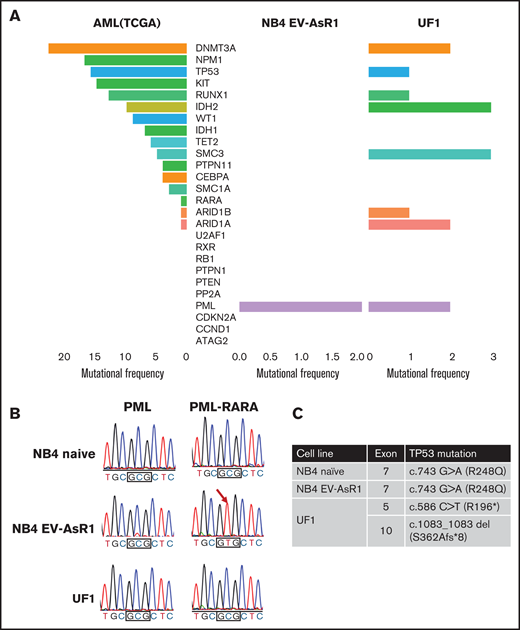
Whole-exome sequencing reveals changes in the ATO-resistant cell lines at the genomic level. (A) The Cancer Genome Atlas (TCGA) AML data set was compared with novel mutations observed in NB4EV-AsR1 and UF1 cell lines; the graph represents the novel mutations (found only in resistant cell lines) and their mutation frequency. (B) Sanger sequencing showing the existence of PMLA216V in the in-house–generated ATO-resistant cell line but not in the UF1 and parental NB4 naïve cell line. (C) Mutations observed in the p53 gene of ATO-resistant and sensitive cell lines.
Whole-exome sequencing reveals changes in the ATO-resistant cell lines at the genomic level. (A) The Cancer Genome Atlas (TCGA) AML data set was compared with novel mutations observed in NB4EV-AsR1 and UF1 cell lines; the graph represents the novel mutations (found only in resistant cell lines) and their mutation frequency. (B) Sanger sequencing showing the existence of PMLA216V in the in-house–generated ATO-resistant cell line but not in the UF1 and parental NB4 naïve cell line. (C) Mutations observed in the p53 gene of ATO-resistant and sensitive cell lines.
Validation of PML and p53 mutations using Sanger sequencing confirmed the existence of previously reported ATO resistance-conferring mutation A216V10 in the in-house–generated ATO-resistant cell lines (including NB4EV-AsR2 and NB4EV-AsR3). UF1 had 2 intronic variations in the PML domain and was negative for A216V (Figure 3B). We also noted the existence of a p53 gain-of-function mutation (R248Q) in the in-house–generated ATO-resistant cell line that was also present in the parental naïve NB4 cell line. The UF1 cell line had a point mutation (R196*) and a deletion of exon 10 of p53 (Figure 3C), which are reported to be pathogenic.20
Heterogeneity in ATO-resistant cell lines
As is evident from drug withdrawal conditions and exome sequencing analysis, the observed ATO resistance was not explained by either a transient epigenetic poising or presence of a known acquired genetic mutation in the PML-B2 domain (absence of PML-B2 domain mutation in UF1). We next subjected NB4 naïve, NB4EV-AsR1, and UF1 cell lines to gene expression profiling. We observed that 1717 genes in NB4EV-AsR1 and 6149 genes in UF1 were significantly upregulated (>twofold) compared with the NB4 naïve cell line. The pathways significantly enriched for differentially expressed genes were cell survival, cell cycle, immune regulation, ATP-binding cassette transporters, glutathione metabolism, the redox system, mitochondrial cellular respiration, and the ubiquitin-proteasome degradation system (Figure 4A). We also noted that the gene expression profile of the in-house–generated ATO-resistant cell line was similar to that of the relapsed APL patient’s gene profile (8 unmatched newly diagnosed and relapsed APL) treated with first-line ATO-based regimens that we had previously reported21 (Figure 4B). Gene expression profiling revealed significant dysregulation of glycolytic and mitochondrial metabolism in the resistant cell line when compared with the NB4 naïve cell line (Figure 4C).
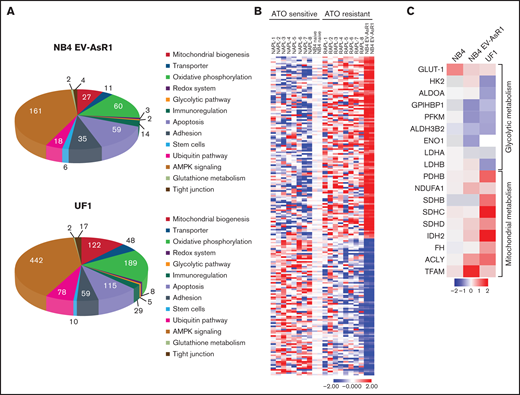
Gene expression analysis of the ATO-resistant cell lines reveals dysregulation of cellular metabolism. (A) Pie chart representing the dysregulated genes in the in-house–generated ATO-resistant NB4EV-AsR1 and UF1 cell lines compared with parental NB4 naïve cell line. (B) Heatmap highlighting gene signature of NB4 naïve (duplicate) in-house–generated ATO-resistant cell line (duplicate) and the 8 unmatched samples from patients with newly diagnosed and relapsed APL (primary cell data was previously reported and was adapted from Chendamari et al).21 (C) Heatmap representing NB4 naïve, ATO-resistant NB4EV-AsR1, and UF1 cell line genes involved in glycolytic and mitochondrial metabolism.
Gene expression analysis of the ATO-resistant cell lines reveals dysregulation of cellular metabolism. (A) Pie chart representing the dysregulated genes in the in-house–generated ATO-resistant NB4EV-AsR1 and UF1 cell lines compared with parental NB4 naïve cell line. (B) Heatmap highlighting gene signature of NB4 naïve (duplicate) in-house–generated ATO-resistant cell line (duplicate) and the 8 unmatched samples from patients with newly diagnosed and relapsed APL (primary cell data was previously reported and was adapted from Chendamari et al).21 (C) Heatmap representing NB4 naïve, ATO-resistant NB4EV-AsR1, and UF1 cell line genes involved in glycolytic and mitochondrial metabolism.
We also carried out a limited analysis of epigenetic modifications using chromatin immunoprecipitation combined with high-throughput sequencing (ChIP-seq) for H3k27ace as an epigenetic marker of active enhancers and promoters and for H3k27me3 as a marker for gene repression. In the NB4EV-AsR1 in-house–generated resistant cell line, gene ontology enrichment on the H3k27ace marker of promoter regions showed significant enrichment for DNA damage, mitochondria, cell cycle, and messenger RNA splicing clusters compared with the naïve NB4 cell line (supplemental File 2).
We validated the finding by measuring the basal metabolic properties such as reactive oxygen species (ROS), antioxidant level, glucose uptake, and mitochondrial membrane potential, which are reported to be key factors in the mechanisms of ATO action. We observed that compared with naïve NB4 cells, ATO-resistant cell lines (NB4EV-AsR1 and UF1) had low levels of basal ROS (Figure 5A), lower mitochondrial membrane potential, and low glucose uptake capacity (2-NBDG uptake, GLUT-1, and LDHA at both transcript and protein levels) compared with the naïve NB4 cells (Figure 5C-E). As expected, glutathione levels were elevated in NB4EV-AsR1 compared with naïve NB4 cells, although it remained low in UF1 cells (Figure 5B). Consistent with the above observation compared with newly diagnosed primary APL samples, we observed significant downregulation of GLUT-1 and LDHA transcript levels in the relapsed APL samples from patients (supplemental Figure 6).
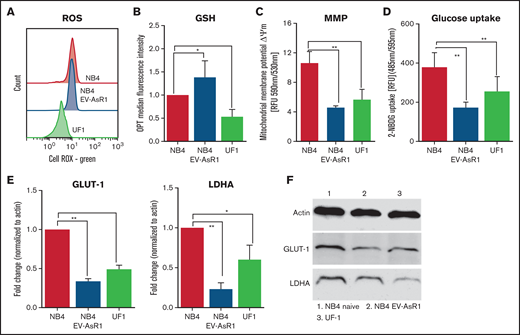
ATO-resistant cell lines are metabolically distinct. (A) Baseline total ROS were measured using redox-sensitive dye (cell ROX Green) in flow cytometry. (B) Bar graphs show baseline protein thiols indicative of antioxidants measured by using o-phthaldialdehyde (OPT) and median fluorescence intensity. (C) Mitochondria membrane potential (MMP) of the resistant cell lines was measured using JC-1. (D) Glucose uptake was measured using a fluorescent analog of 2-DG and is represented as relative mean fluorescence intensity. GLUT-1 and LDHA transcripts (E) and protein levels of NB4 naïve (F), NB4EV-AsR1, and UF1 cell lines. All error bars represent mean ± SEM for 3 to 4 independent experiments. *P ≤ .05; **P ≤ .01. GSH, glutathione; RFU, relative fluorescence units.
ATO-resistant cell lines are metabolically distinct. (A) Baseline total ROS were measured using redox-sensitive dye (cell ROX Green) in flow cytometry. (B) Bar graphs show baseline protein thiols indicative of antioxidants measured by using o-phthaldialdehyde (OPT) and median fluorescence intensity. (C) Mitochondria membrane potential (MMP) of the resistant cell lines was measured using JC-1. (D) Glucose uptake was measured using a fluorescent analog of 2-DG and is represented as relative mean fluorescence intensity. GLUT-1 and LDHA transcripts (E) and protein levels of NB4 naïve (F), NB4EV-AsR1, and UF1 cell lines. All error bars represent mean ± SEM for 3 to 4 independent experiments. *P ≤ .05; **P ≤ .01. GSH, glutathione; RFU, relative fluorescence units.
ATO-resistant APL cell lines are metabolically distinct compared with ATO-sensitive cell lines
A glyco stress test revealed that the naïve NB4 cells had increased glycolysis in which the basal extracellular acidification rate was significantly higher (Figure 6A) with increased oxygen consumption rate (supplemental Figure 7) compared with ATO-resistant cell lines. To address the degree to which glycolysis is necessary for cell survival, we treated the naïve NB4 and ATO-resistant cell lines with 2-DG, a glucose analog that inhibits glycolysis via its action on hexokinases. We noted that the viability of the naïve NB4 cell line was significantly affected in the presence of 2-DG, equivalent to the effect seen with 2 µM of ATO. There was no evidence of an additive effect when these agents were combined (Figure 6B). In contrast, the viability of the APL-resistant cell lines and AML cell lines (U937 and THP-1; data not shown) was not significantly affected when 2-DG was used alone or in combination with ATO (Figure 6B).
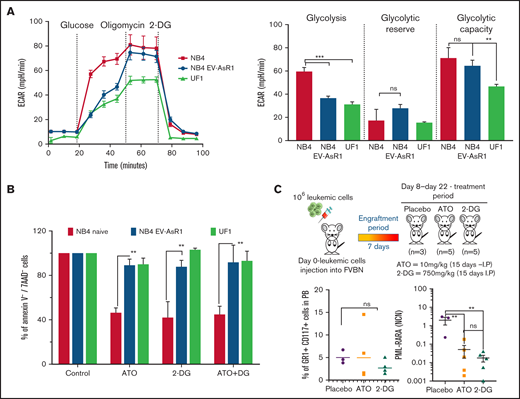
ATO-resistant cells are metabolically heterogeneous, and the in vivo effect of glycolytic inhibition by 2-DG reduces leukemic burden in the APL mouse model. (A) The extracellular acidification rate (ECAR) and glycolytic potential of NB4 naïve, NB4EV-AsR1, and UF1 cell lines were assessed in real time by using a Seahorse extracellular flux analyzer. (B) Viability of the sensitive and resistant cell lines after 48 hours of glycolytic inhibitor ATO and 2-DG (ATO, 2 µM; 2-DG, 5 mM; n = 4). (C) Schematic representation of the APL transplantable mouse model and treatment plan. Mice were euthanized on day 22 and examined for the presence of leukemic cells (CD117+Gr1+) in peripheral blood (PB) and PML-RARA transcript levels in bone marrow. All error bars represent mean ± SEM of 4 independent experiments. **P ≤ .01; ***P ≤ .001. IP, intraperitoneal; ns, not significant.
ATO-resistant cells are metabolically heterogeneous, and the in vivo effect of glycolytic inhibition by 2-DG reduces leukemic burden in the APL mouse model. (A) The extracellular acidification rate (ECAR) and glycolytic potential of NB4 naïve, NB4EV-AsR1, and UF1 cell lines were assessed in real time by using a Seahorse extracellular flux analyzer. (B) Viability of the sensitive and resistant cell lines after 48 hours of glycolytic inhibitor ATO and 2-DG (ATO, 2 µM; 2-DG, 5 mM; n = 4). (C) Schematic representation of the APL transplantable mouse model and treatment plan. Mice were euthanized on day 22 and examined for the presence of leukemic cells (CD117+Gr1+) in peripheral blood (PB) and PML-RARA transcript levels in bone marrow. All error bars represent mean ± SEM of 4 independent experiments. **P ≤ .01; ***P ≤ .001. IP, intraperitoneal; ns, not significant.
Having noted that glycolytic inhibition by 2-DG promoted apoptosis in NB4 cells comparable to that seen with ATO, we performed in vivo glycolytic inhibition to understand its physiological relevance in the ATO-sensitive transplantable APL mouse model. We observed that 2-DG or ATO as single agents reduced the leukemic burden in the peripheral blood and PML-RARA copy number at the end of 22 days to levels that were comparable and indistinguishable from each other (Figure 6C).
Heterogeneity of anti-apoptotic protein dependency of ATO-resistant APL cell lines
We next assessed the mitochondria priming status of the ATO-resistant cell lines by using iBH3 profiling and their sensitivity to the BH3 mimetics. We observed a significant difference between the in-house ATO-resistant cell lines and the parental cell line (independent of A216V). Neither a BCL-2 inhibitor (ABT-199; venetoclax) nor a BCL-XL inhibitor (A1331852) promoted apoptosis as a single agent in the parental and resistant cell lines, whereas an MCL-1 inhibitor (S63845) promoted apoptosis only in parental NB4 naïve and NB4EV-AsR2 cell lines (Figure 7A). The sensitivity to the BH3 mimetics correlated with their basal BH3 profiling (Figure 7B).
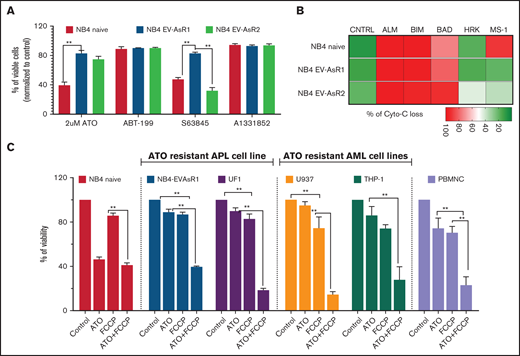
Mitocans synergize with ATO to promote apoptosis in the ATO-resistant cell lines. (A) Viability of NB4 and in-house–generated ATO-resistant cell lines to BH3 mimetics (n = 5). (B) Intracellular BH3 profiling of ATO-resistant and sensitive cells (n = 3) measured using intracellular cytochrome-c (Cyto-C) retention. (C) Viability of the sensitive and resistant APL and non-APL cell lines treated for 48 hours with OXPHOS uncoupler FCCP combined with ATO (ATO, 2 µM; FCCP, 10 µM; BH3 mimetics, 250 nM). All error bars represent the mean ± SEM of 4 independent experiments. *P ≤ .05; **P ≤ .01; ***P ≤ .001; ***P ≤ .0001. A1331852, BCL-XL inhibitor; ABT-199, venetoclax; CNTRL, control; S63845, MCL-1 inhibitor.
Mitocans synergize with ATO to promote apoptosis in the ATO-resistant cell lines. (A) Viability of NB4 and in-house–generated ATO-resistant cell lines to BH3 mimetics (n = 5). (B) Intracellular BH3 profiling of ATO-resistant and sensitive cells (n = 3) measured using intracellular cytochrome-c (Cyto-C) retention. (C) Viability of the sensitive and resistant APL and non-APL cell lines treated for 48 hours with OXPHOS uncoupler FCCP combined with ATO (ATO, 2 µM; FCCP, 10 µM; BH3 mimetics, 250 nM). All error bars represent the mean ± SEM of 4 independent experiments. *P ≤ .05; **P ≤ .01; ***P ≤ .001; ***P ≤ .0001. A1331852, BCL-XL inhibitor; ABT-199, venetoclax; CNTRL, control; S63845, MCL-1 inhibitor.
Combination of glycolytic inhibitor (ATO) and mitocans promoted apoptosis in the ATO-resistant cell lines
We then evaluated the effect of a mitochondrial oxidative phosphorylation (OXPHOS) uncoupler on the ATO-resistant promyelocytic and non-promyelocytic AML cell lines. The viability of ATO-resistant cell lines was not affected significantly when treated with the mitochondrial OXPHOS uncoupler FCCP as a single agent, but when it was combined with ATO, the viability was significantly reduced in NB4EV-AsR1 cells (harbors A216V mutation; resistant to BCL-2 and MCL-1 inhibition), NB4EV-AsR2 cells (harbors A216V mutation and is sensitive to MCL-1 inhibition), and the UF1 cell line (A216V negative, sensitive to BCL-2 and MCL-1 inhibition) (Figure 7C). A similar effect was observed in the non-M3 AML U937 and THP-1 cell lines (resistant to BCL-2 inhibition and ATO but sensitive to MCL-1; supplemental Figure 8). We observed that the ATO-resistant APL and AML cell lines were significantly different in their metabolic preferences for survival from the ATO-sensitive NB4 naïve cell line, and by targeting this difference, we were able to overcome the resistance independently of the existence of the PML-B2 domain mutation and its anti-apoptotic protein dependency. Therefore, blocking mitochondrial respiration in combination with ATO can enhance cell death in ATO- and ATRA-resistant APL cell lines and non-M3 AML cell lines. However, this combination had significant off-target effects on the normal peripheral blood mononuclear cells, and further evaluation of compounds that selectively target the leukemic cell’s mitochondrial respiration is needed before these observations can be translated to the clinic.
Discussion
The study highlights that, compared with the existing ATO-resistant APL cell lines,22,23 our in-house–generated ATO-resistant cell lines are stable and well-characterized at the genomic levels and they also possess the well-known ATO resistance-conferring mutation A216V in the B2 domain of the PML-RARA oncoprotein. These ATO-resistant cell lines were observed to be less sensitive to differentiation with ATRA and to therapy with the combination of ATO and ATRA; thus, they are an excellent tool for evaluating additional mechanisms that could contribute to drug resistance. We also noted that the UF1 APL cell line was resistant to ATO (this has not been previously reported) but did not have the well-defined PML-RARA B2 domain mutation. This gave us an opportunity to study ATO resistance with and without PML-RARA B2 domain mutations.
Compared with the naïve NB4 cell line, in-house–generated ATO-resistant cell lines overexpressed ATO efflux transporters such as AQP9, MRP4, and ABCA7, which correlated with their inability to accumulate intracellular ATO. This phenomenon was not observed in primary blasts from patients with relapsed APL who had previously been treated with ATO compared with newly diagnosed patients.21 The possibility of efflux transporters as a protective mechanism of ATO resistance cannot be excluded in the clinic and needs further evaluation.
We noted the presence of a somatic mutation in the B2 domain of the PML gene in our in-house–generated ATO-resistant cell line that has been reported to confer resistance to ATO. It is important to note that these mutations are acquired after ATO treatment, and none of the APL cell lines or patients with relapsed APL before ATO therapy had the B2 domain mutation.24 The in-house resistant cell lines did not acquire additional mutations in the p53 gene, which has been reported to be critical for regulating the formation of NB and generation of ROS. Our observation suggests that the reduction in PML micro-speckled pattern formation and low ROS levels are independent of p53 status in these cell lines. The cellular redox system of the resistant cell lines was found to be significantly altered with lower ROS levels, lower proliferative rate, and an increased antioxidant system that favored quiescence and stemness-like properties.25 This was further corroborated by the observation of low extracellular acidification rate and oxygen consumption rate status of the resistant cell lines.26
The NB4 naïve ATO-sensitive cell line was more reliant on the Warburg effect for survival and proliferation, and its viability was significantly affected by a glycolytic inhibitor (2-DG). In an in-vivo APL model, 2-DG significantly reduced the leukemic burden comparable to the standard of care (ATO). This further supports our observation that the naïve ATO-sensitive APL cells’ survival could be targeted by glycolytic inhibition. On the basis of the recognized inhibitory effect of ATO on the glycolytic pathway,17 this could also be an important mechanism by which ATO induces apoptosis in malignant promyelocytes. In contrast, the ATO-resistant APL and AML cell lines were mainly dependent on OXPHOS for their survival. However, unlike NB4 naïve cells, the ATO-resistant and AML cell lines had greater metabolic plasticity to switch between glycolysis and OXPHOS when one is inhibited but were susceptible to a combination of ATO and mitocans. This susceptibility was independent of their PML B2 domain mutation status and their anti-apoptotic protein dependency. It is well known that A216V mutation alters the ATO binding on the B2 domain of the PML-RARA oncoprotein. The observed effect of ATO combined with mitocans on the resistant cell lines is thus likely to be a result of the inhibitory effect of ATO on the glycolytic pathway.13,27
Taken together, our work demonstrates that ATO resistance is multifactorial and is not limited to the presence or absence of either PML or p53 mutations. The in-house–generated ATO-resistant cell line would be a useful model for evaluating mechanisms of resistance in leukemia. Targeting the metabolic adaptations seen in ATO-resistant cell lines has the potential to overcome such resistance. Inhibiting mitochondrial respiration combined with ATO could overcome ATO resistance, and translating this approach to the clinic for treating patients with relapsed APL and those who are newly diagnosed needs to be explored further. Although the combination of ATO and FCCP was observed to be nonselective, there are a number of drugs used in the clinic (approved by the US Food and Drug Administration) that can selectively inhibit mitochondrial respiration in cancer cells. These could be evaluated for their synergistic activity with ATO in leukemia.28 Our data also draw attention to possible severe off-target toxicity of such combinations that may be inadvertently used in the clinic.
Acknowledgments
The authors thank Intas Pharmaceutical (India) and NATCO Pharmaceutical (India) for providing active pharmaceutical ingredients of pharmaceutical drugs for this study, Mitradas Panicker, PhD, National Center for Biological Sciences (Bengaluru, India) and Tamil Selvan, PhD, Department of Biotechnology, Anna University (Chennai, India) for providing access to the seahorse extracellular flux analyzer.
This study was supported by grants from India Alliance Wellcome-Department of Biotechnology in India (DBT) (IA/CPHS/18/1/503930), Department of Biotechnology-Centre of Excellence (DBT-COE) (BT/COE/34/SP13432/2015), and Department of Science and Technology-Science and Engineering Research Board (DST-SERB) (CRG/2019/001214) (New Delhi, India). V.M. was supported by the senior fellowship program of India Alliance Wellcome-DBT (IA/CPHS/18/1/503930) (New Delhi, India). S.G., H.K.P., and S.D. were supported by senior research fellowships from the Council for Scientific and Industrial Research (New Delhi, India). A.V. was supported by a junior research fellowship from DBT, Government of India. The Seahorse facility at Anna University is funded by the Department of Science and Technology Fund for Improvement of Science and Technology (S&T) Infrastructure (SR/FST/LSI-649/2015).
Authorship
Contribution: N.B. helped design the study, performed research and molecular tests, analyzed data, and helped write the paper; S.G., E.C., H.K.P., A.V., A.A.A., S.D., S.P.K., and N.R.R. performed research and molecular tests and analyzed data; M.Y.M., and N.B.J. performed research and karyotyping tests and analyzed data; A.K., U.K., S.K., and P.B. performed research and analyzed data; and V.M. helped design the study, performed research, analyzed data, and helped write the paper.
Conflict-of-interest disclosure: The authors declare no competing financial interests
Correspondence: Vikram Mathews, Department of Haematology, Christian Medical College, IDA Scudder Rd, Vellore 632004, Tamil Nadu, India; e-mail: vikram@cmcvellore.ac.in.

