

TO THE EDITOR:
Sequence variant interpretation (SVI) is a formal process by which gene variants are classified as pathogenic (P), likely pathogenic (LP), variant of uncertain significance, likely benign (LB), or benign (B). The classifications, based on the 28 criteria defined by the American College of Medical Genetics and Genomics (ACMG) and Association for Molecular Pathology (AMP),1 are intended to undergo gene-specific modification facilitated by different Clinical Genome Resource (ClinGen) Variant Curation Expert Panels (VCEPs). The ClinGen Myeloid Malignancy Variant Curation Expert Panel (MM-VCEP) is charged with developing curation rules for genes in which variants confer risk for myeloid malignancies, starting with RUNX1.2,3 To reflect the continual updates from current literature and ClinVar submissions accurately, ClinGen advocates reevaluation of variant curation specifications on a regular basis. In response to this recommendation coupled with additional refinement of ACMG/AMP criteria by the ClinGen SVI Working Group, the MM-VCEP has updated its RUNX1 curation rules to increase accuracy of curation in the fields of hematology, genetics, and pathology while optimizing clinical assessments of patients undergoing testing for inherited predisposition (Table 1; Figure 1A; supplemental case studies). With initial RUNX1 criteria being published in 2019, the reevaluation in 2021 lent itself to the update of 7 criteria: PM1, PM2, PM4, PM5, PP3, BP4, and BP7. Here, we outline the revised RUNX1 curation rules, now designated as version 2 (supplemental Table 1).
Updated RUNX1-specific variant curation rules
| ACMG/AMP criteria code | RUNX1-specification version 1 | ACMG/AMP criteria code | RUNX1-specification version 2 |
|---|---|---|---|
| PM1_supporting | Missense variant affecting an aa residue between aa residues 105 and 204 within the RHD | PM1_supporting | Missense variant affecting an aa residue between aa residues 89 and 204 within the RHD |
| PM2 | Variant must be completely absent from all population databases. The mean coverage of RUNX1 in the population database used should be at least 20× | PM2_supporting | Variant must be completely absent from all population databases. The mean coverage of RUNX1 in the population database used should be at least 20× Note: PM2 is downgraded to PM2_supporting in this version |
| PM4_supporting | In-frame deletion/insertion affecting residues 105-204 within the RHD | PM4_supporting | In-frame deletion/insertion affecting residues 89-204 within the RHD |
| PM5_supporting | Missense change at the same residue where a different missense change has previously been determined to be LP | PM5_supporting | Applicable for: 1. Missense change at the same residue where a different missense change has previously been determined to be LP 2. Nonsense/frameshift variants that are downstream of c.98 (in transcript NM_001754.5) |
| PP3 | Applicable for: 1. Missense variants with a REVEL score >0.75 2. Missense or synonymous variants if the variant alters the last 3 bases of an exon preceding a donor splice site or the first 3 bases of an exon following a splice acceptor site and the predicted decrease in the score of the canonical splice site (measured by both MES and SSF-like) is at least 75% regardless of the predicted creation/presence of a putative cryptic splice site 3. Intronic variants (in introns 4-8) located in reference to exons at positions +3 to +5 for splice donor sites or −3 to −5 for splice acceptor sites for which the predicted decrease in the score is at least 75% (measured by both MES and SSF-like) regardless of the predicted creation/presence of a putative cryptic splice site Note: PP3 cannot be applied for canonical splice site variants | PP3 | Applicable for: 1. Missense variants with a REVEL score ≥0.88 and/or a SpliceAI score ≥0.38 2. Synonymous and intronic variants with any splicing prediction in SpliceAI ≥0.38. Note: PP3 cannot be applied for canonical splice site variants or 5′/3′ UTR variants |
| BP4 | Applicable for missense variants if all the following apply: 1. REVEL score <0.15 2. MES and SSF-like predict either an increase in the canonical splice site score or a decrease in the canonical splice site score by no more than 10% 3. No putative cryptic splice sites are created Applicable for synonymous, intronic, and noncoding variants if MES and SSF-like predict either an increase in the canonical splice site score or a decrease in the canonical splice site score by no more than 10%, and no putative cryptic splice sites are created | BP4 | Applicable for: 1. Missense variants with a REVEL score ≤0.50 and SpliceAI score ≤0.20 2. Synonymous and intronic variants with any splicing prediction in SpliceAI score ≤0.20 |
| BP7 | Applicable for intronic/noncoding variants at or beyond positions +7/–21 for which MES and SSF-like predict either an increase in the canonical splice site score or a decrease in the canonical splice site score by no more than 10%, and no putative cryptic splice sites are created. Additionally requires that evolutionary conservation tools predict the nucleotide is not conserved (ie, phyloP score ≤0.1 or the variant allele is the reference nucleotide in 1 primate and/or 3 mammal species) | BP7 | Applicable for synonymous and intronic variants for which the SpliceAI score ≤0.20 AND evolutionary conservation tools predict the site is not highly conserved (ie, phyloP100way score in GRCh38/hg38 ≤2.0 and/or the variant allele is the reference nucleotide in 1 primate and/or 3 mammal species) Note: BP7 cannot be applied for 5′/3′ UTR variants |
| ACMG/AMP criteria code | RUNX1-specification version 1 | ACMG/AMP criteria code | RUNX1-specification version 2 |
|---|---|---|---|
| PM1_supporting | Missense variant affecting an aa residue between aa residues 105 and 204 within the RHD | PM1_supporting | Missense variant affecting an aa residue between aa residues 89 and 204 within the RHD |
| PM2 | Variant must be completely absent from all population databases. The mean coverage of RUNX1 in the population database used should be at least 20× | PM2_supporting | Variant must be completely absent from all population databases. The mean coverage of RUNX1 in the population database used should be at least 20× Note: PM2 is downgraded to PM2_supporting in this version |
| PM4_supporting | In-frame deletion/insertion affecting residues 105-204 within the RHD | PM4_supporting | In-frame deletion/insertion affecting residues 89-204 within the RHD |
| PM5_supporting | Missense change at the same residue where a different missense change has previously been determined to be LP | PM5_supporting | Applicable for: 1. Missense change at the same residue where a different missense change has previously been determined to be LP 2. Nonsense/frameshift variants that are downstream of c.98 (in transcript NM_001754.5) |
| PP3 | Applicable for: 1. Missense variants with a REVEL score >0.75 2. Missense or synonymous variants if the variant alters the last 3 bases of an exon preceding a donor splice site or the first 3 bases of an exon following a splice acceptor site and the predicted decrease in the score of the canonical splice site (measured by both MES and SSF-like) is at least 75% regardless of the predicted creation/presence of a putative cryptic splice site 3. Intronic variants (in introns 4-8) located in reference to exons at positions +3 to +5 for splice donor sites or −3 to −5 for splice acceptor sites for which the predicted decrease in the score is at least 75% (measured by both MES and SSF-like) regardless of the predicted creation/presence of a putative cryptic splice site Note: PP3 cannot be applied for canonical splice site variants | PP3 | Applicable for: 1. Missense variants with a REVEL score ≥0.88 and/or a SpliceAI score ≥0.38 2. Synonymous and intronic variants with any splicing prediction in SpliceAI ≥0.38. Note: PP3 cannot be applied for canonical splice site variants or 5′/3′ UTR variants |
| BP4 | Applicable for missense variants if all the following apply: 1. REVEL score <0.15 2. MES and SSF-like predict either an increase in the canonical splice site score or a decrease in the canonical splice site score by no more than 10% 3. No putative cryptic splice sites are created Applicable for synonymous, intronic, and noncoding variants if MES and SSF-like predict either an increase in the canonical splice site score or a decrease in the canonical splice site score by no more than 10%, and no putative cryptic splice sites are created | BP4 | Applicable for: 1. Missense variants with a REVEL score ≤0.50 and SpliceAI score ≤0.20 2. Synonymous and intronic variants with any splicing prediction in SpliceAI score ≤0.20 |
| BP7 | Applicable for intronic/noncoding variants at or beyond positions +7/–21 for which MES and SSF-like predict either an increase in the canonical splice site score or a decrease in the canonical splice site score by no more than 10%, and no putative cryptic splice sites are created. Additionally requires that evolutionary conservation tools predict the nucleotide is not conserved (ie, phyloP score ≤0.1 or the variant allele is the reference nucleotide in 1 primate and/or 3 mammal species) | BP7 | Applicable for synonymous and intronic variants for which the SpliceAI score ≤0.20 AND evolutionary conservation tools predict the site is not highly conserved (ie, phyloP100way score in GRCh38/hg38 ≤2.0 and/or the variant allele is the reference nucleotide in 1 primate and/or 3 mammal species) Note: BP7 cannot be applied for 5′/3′ UTR variants |
GRCh38/hg38, Genome Reference Consortium Human Build 38; MES, MaxEntScan; RHD, runt homology domain; SSF-like, splice site finder-like; UTR, untranslated region.
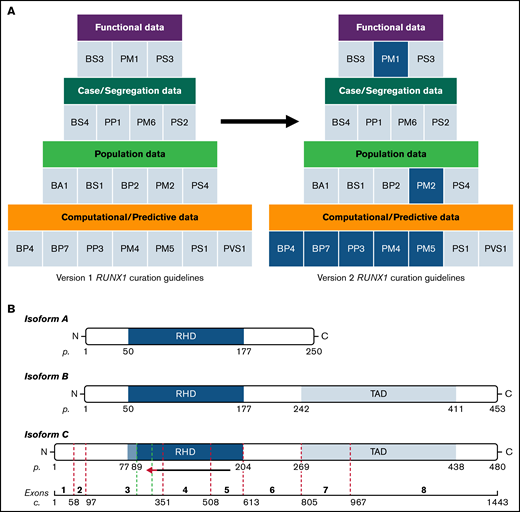
RUNX1 variant curation codes and isoform schematic. (A) The updated RUNX1 curation codes improve the curation process and increase the accuracy of variant classification. The first version of RUNX1-specific ACMG/AMP codes is shown on the left. The updated RUNX1-specific codes (version 2) are displayed on the right. Only revised codes are highlighted with blue fill. (B) Alternative splicing creates 3 protein isoforms that are shown schematically: A (top), B (middle), and C (bottom, isoform used for variant curation). The exons encoding isoform C are shown at the bottom of the figure. A red arrow indicates the extension of the critical region for variant curation defined by the updated curation rules. The green dotted lines indicate the extension of the amino acid range considered for PM1 and PM4. c, nucleotide numbering; p, amino acid numbering; RHD, RUNT homology domain; TAD, transactivation domain. Nucleotide numbering is according to NM_001754.5.
RUNX1 variant curation codes and isoform schematic. (A) The updated RUNX1 curation codes improve the curation process and increase the accuracy of variant classification. The first version of RUNX1-specific ACMG/AMP codes is shown on the left. The updated RUNX1-specific codes (version 2) are displayed on the right. Only revised codes are highlighted with blue fill. (B) Alternative splicing creates 3 protein isoforms that are shown schematically: A (top), B (middle), and C (bottom, isoform used for variant curation). The exons encoding isoform C are shown at the bottom of the figure. A red arrow indicates the extension of the critical region for variant curation defined by the updated curation rules. The green dotted lines indicate the extension of the amino acid range considered for PM1 and PM4. c, nucleotide numbering; p, amino acid numbering; RHD, RUNT homology domain; TAD, transactivation domain. Nucleotide numbering is according to NM_001754.5.
As recommended by the SVI Working Group, the MM-VCEP will now incorporate the use of a Bayesian framework. Curated RUNX1 variants will be classified based on computed posterior probabilities derived by integrating the applicable ACMG/AMP criteria quantitatively, particularly when such data conflict with benign and pathogenic codes (supplemental Table 2).4,5
The MM-VCEP now follows the SVI recommendation to downgrade the strength of PM2 to PM2_supporting.6
In the original rules, BP7 was applied for synonymous/intronic variants with a PhyloP score7 <0.1. However, this threshold was found to be overly conservative, as it restricted the use of BP7 to nonconserved regions. By obtaining the PhyloP scores for all RUNX1 variants in the Genome Aggregation Database that met population frequency thresholds established for BA1/BS1, a threshold change of the PhyloP score to ≤2.0 was determined to be sufficient to still encompass >85% concordance with the respective B/LB classification among the synonymous and intronic variant sets (supplemental Tables 3 and 4).
Analysis of 25 nonsense/frameshift RUNX1 variants curated by the MM-VCEP classified at least 2 variants as P in exons 3 to 7 and 6 variants in exon 8 as P/LP without the application of PM5_supporting (supplemental Table 5). Thus, in addition to using PM5_supporting for a missense change at an amino acid residue where a different missense change has been determined to be LP, this criterion will now also include nonsense/frameshift variants downstream of c.98. Note that nonsense/frameshift variants upstream of c.98 affect only RUNX1 transcript C (NM_001754.5; Figure 1B), and therefore, this update is not applicable for these variants.
In silico tools are used to predict the effect of missense variants as being neutral (BP4) or deleterious (PP3). The MM-VCEP recommends using REVEL, a meta-predictor that combines 13 individual tools with high sensitivity and specificity with better performance to any individual tool or ensemble method.8-10 In the original RUNX1 guidelines, variants with a REVEL score of <0.15 would allow application of BP4, whereas those with a score >0.75 would allow application of PP3. Over the past several years, additional ensemble in silico tools have been developed. To identify the best performing tool for RUNX1 variants, we computed receiver operating characteristic (ROC) curves and compared the area under the ROC curve (AUC) for 11 different algorithms, using a set of 25 P/LP germline missense variants and 25 LB/B missense variants (supplemental Table 6). BayesDel, Condel, MetaLR, and REVEL performed equally with an AUC of 1 (supplemental Figure 1). As REVEL remains the preferred in silico predictor of the ClinGen SVI working group, new REVEL thresholds were established based on 90% sensitivity for PP3 (REVEL score ≥0.88) and 90% specificity for BP4 (REVEL score ≤0.50).
New computational tools for predicting variant effects on splicing have been developed. The MM-VCEP had recommended using SSF-like in combination with MaxEntScan (MES).2 However, SpliceAI, a new deep-learning based tool, shows an advantage over these and other splice predictors, such as Human Splicing Finder, NNSPLICE, and GeneSplicer, particularly when predicting novel cryptic splice sites and analyzing splice sites positioned further from exon boundaries.11-13 To compare the performance between SpliceAI and MES (previously the best performing splicing predictor evaluated), 202 noncanonical splice variants with functional data in 51 genes associated with inherited hematologic malignancies, aplastic anemia, bone marrow failure syndromes, or inherited cytopenias were considered.14 Of those, 149 variants affected splicing based on published functional data, and 53 were found to have no impact, as based on functional data or by meeting BA1 thresholds. By performing ROC curve analysis, SpliceAI was identified as the superior splice site predictor with an AUC of 0.93 compared with MES, which had an AUC of 0.79.14 Therefore, the MM-VCEP adopted SpliceAI and established thresholds for PP3 with SpliceAI Δ score of ≥0.38, based on 90% sensitivity, and for BP4 and BP7 with SpliceAI Δ score of ≤0.20, based on 90% specificity.
SpliceAI can evaluate deep intronic and exonic splice variants, which allows the removal of the prior positional restriction (ie, +7/−21 in BP7 and ±3/±5 in PP3) that limited evaluation to splice variants located close to intron and exon boundaries. Because splicing algorithms do not work on UTR variants, these are not applied to UTR variants.
PM1_supporting was applied previously to missense variants encoding amino acids 105 to 204. However, the MM-VCEP noted that curation of RUNX1 p.Ser94Arg yielded a designation of LP, without application of PM1_supporting. Moreover, germline variants in RUNX2, a gene with 90% sequence homology to the RUNX1 RUNT Homology Domain, were identified affecting amino acids 89 and 94 and classified as P/LP,15,16 suggesting the amino acid range for PM1_supporting could be extended. Last, the RUNX1 protein structure shows amino acid 89 as the start of the β-sheet portion of the core binding factor β heterodimerization domain, noted as functionally important.3,17 Therefore, the MM-VCEP extended the amino acid range to 89 to 204 for codes PM1_supporting and PM4_supporting.
These revisions to RUNX1 variant curation rules provide an important resource for hematology, genetics, and pathology communities that will facilitate accuracy of variant pathogenicity, which is critical for patient care and follow-up. We look forward to the future application of new tools as a means of continuing to improve gene variant curation. Tools utilizing predictive models, such as deep generative models like Evolutionary model of Variant Effect (EVE)18 and functional testing19,20 may allow us to identify and prioritize variants more accurately.
Acknowledgments: The MM-VCEP is supported by the National Institutes of Health, National Cancer Institute grant 1U24CA258118-01. S.F. receives funding from the Olympia Morata Program of the Medical Faculty Heidelberg.
Contribution: S.F., X.L., M.S., T.W., N.M., D.W., and L.A.G. contributed to the conception and design of the work, drafted the manuscript, revised it critically, gave approval for the final version to be published, and agreed to be accountable for all aspects of the work.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
A complete list of the members of the Myeloid Malignancy Variant Curation Expert Panel appears in “Appendix.”
Correspondence: David Wu, 825 Eastlake Ave E, SCCA Room G-7800, Seattle, WA 98109; e-mail: dwu2@uw.edu; and Lucy A. Godley, 5841 S Maryland Ave, MC 2115, Chicago, IL 60637; e-mail: lgodley@medicine.bsd.uchicago.edu.
Appendix: panel members
The Myeloid Malignancy Variant Curation Expert Panel consists of Anupriya Agarwal, Panagiotis Baliakas, Piers Blombery, Emery Bresnick, Anna Brown, Katherine Calvo, Michael Chicka, Jean Donadieu, Simone Feurstein, Christopher Hahn, Claire Homan, Amy Hsu, Sarah Jackson, Siobhan Keel, Chimene Kesserwan, Sarah King-Smith, Lise Larcher, Vincent-Phillippe Lavallée, Zejuan Li, Minjie Luo, Xi Luo, Emily Mace, Luca Malcovati, Shannon McWeeney, Nikita Mehta, Shruthi Mohan, Kim Nichols, Daniel Pineda, Christopher Porter, Mancy Shah, Jean Soulier, Nancy Speck, Eran Tallis, Parvathy Venugopal, Tom Vulliamy, Taylor Walker, Michael Walsh, Ying Wang, Marcin Wlodarski, David Wu, and Liying Zhang.

