

Key Points
The expression of NMII-A E1841K mutant in endothelial cells impairs VWF secretion induced by cAMP signaling.
Endothelium-specific Myh9 E1841K mutant mice exhibit a prolonged bleeding time with normal platelets.

Abstract
Mutations in MYH9, the gene encoding the heavy chain of nonmuscle myosin IIa (NMII-A), cause MYH9-related disease (MYH9-RD), which is an autosomal-dominant thrombocytopenia with bleeding tendency. Previously, we showed that NMII-A in endothelial cells (ECs) is critical for hemostasis via regulating von Willebrand factor (VWF) release from Weibel-Palade bodies (WPBs). The aim of this study was to determine the role of the expression of MYH9 mutants in ECs in the pathogenesis of the MYH9-RD bleeding symptom. First, we expressed the 5 most common NMII-A mutants in ECs and found that E1841K mutant-expressing ECs secreted less VWF than the controls in response to a cyclic adenosine monophosphate (cAMP) signaling agonist. Then, we generated 2 knockin mouse lines, 1 with Myh9 E1841K in ECs and the other in megakaryocytes. Endothelium-specific E1841K mice exhibited impaired cAMP-induced VWF release and a prolonged bleeding time with normal platelets, whereas megakaryocyte-specific E1841K mice exhibited macrothrombocytopenia and a prolonged bleeding time with normal VWF release. Finally, we presented mechanistic findings that E1841K mutation not only interferes with S1943 phosphorylation and impairs the peripheral distribution of Rab27a-positive WPBs in Ecs under quiescent condition but also interferes with S1916 phosphorylation by disrupting the interaction with zyxin and CKIIα and reduces actin framework formation around WPBs and subsequent VWF secretion under the stimulation by a cAMP agonist. Altogether, our results suggest that impaired cAMP-induced endothelial VWF secretion by E1841K mutant expression may contribute to the MYH9-RD bleeding phenotype.
Introduction
Nonmuscle myosin Iia (NMII-A), one of the isoforms of non-muscle myosin II heavy chain encoded by MYH9 gene, contains a globular head region with an MgATP binding domain, an α-helical coiled coil, and a nonhelical tail. Mutations in this gene are linked to an autosomal dominant disorder called MYH9-related disease (MYH9-RD).1-3 In ∼80% of the affected families, MYH9-RD mutations are concentrated in 6 amino-acids: S96 (6%) and R702 (24%) in the head domain; R1165 (9%), D1424 (20%), and E1841 (22%) in the coiled coil; and R1933 (19%) in the nonhelical tail.4 Patients exhibit congenital macrothrombocytopenia and a bleeding tendency,4-8 which have been phenocopied in MYH9-RD mouse models.9 So far, mechanistic studies of the bleeding symptom have focused on platelet defects, including developmental (such as decreased number and increased size of proplatelets and platelets) and functional (such as a prolonged bleeding time and decreased ability to retract clots) abnormalities in MYH9-RD patients and mouse models.10-12 Although NMII-A is widely expressed,2 no additional major extraplatelet pathologies have been identified for the bleeding tendency of patients with MYH9-RD.
NMII-A is strongly expressed in endothelial cells (Ecs)13 and has several biological functions, such as the maintenance of endothelial barrier integrity and the regulation of cell adhesion and migration in angiogenesis.14-20 NMII-A also plays an important role in both the in vitro and in vivo release of von Willebrand factor (VWF), a multimeric adhesive glycoprotein, from human and mouse Ecs.13 In the quiescent condition, NMII-A with phosphorylated S1943 maintains the normal peripheral distribution of mature Weibel-Palade bodies (WPBs). On stimulation of cyclic adenosine monophosphate (cAMP) agonists, NMII-A is phosphorylated at S1916 and promotes the formation of a zyxin-mediated actin framework around WPBs before fusion, facilitating cAMP-mediated WPB exocytosis.13 Because VWF mediates the adhesion of platelets to components of the extracellular matrix21 and prevents the clearance of procoagulant factor from plasma,22 endothelial NMII-A may be required for thrombosis after vascular injury. Indeed, NMII-A–deficient mice exhibit hindered epinephrine-stimulated VWF release, a prolonged bleeding time, and disrupted thrombosis.13
Given its role in hemostasis, it has been proposed that disruption of NMII-A–mediated cAMP-induced endothelial VWF secretion by expression of MYH9-RD mutants may contribute to the bleeding phenotype.23 Here, we report that endothelial NMII-A E1841K mutant impairs cAMP-induced VWF secretion both in vitro and in vivo and contributes to the bleeding phenotype in MYH9-RD mouse models.
Methods
Cell cultures
Human umbilical vascular endothelial cells (HUVECs) were isolated and cultured as previously described.24 Mouse primary endothelial cells were isolated from adult mouse brain. In brief, brain tissue was minced with surgical blades and then dissolved in serum-free Dulbecco’s Modified Eagle’s Medium (DMEM) containing 1.05 mg/mL of type II collagenase and 58.5 U/mL of type I DNase for 60 minutes at 37°C. The homogenate was pelleted by centrifugation (1000g, 10 minutes, 4°C), resuspended in DMEM containing 20% BSA, and centrifuged (1000g, 20 minutes, 4°C) to separate myelin. The pellet was resuspended in 1 mL of cold DMEM, overlaid on a cold 33% continuous isotonic Percoll gradient, and centrifuged (1000g, 10 minutes, 4°C). The brain EC (BEC) layer (near the bottom) was collected and washed in cold DMEM by centrifugation (1000g, 5 minutes, 4°C). BECs were cultured as for HUVECs.25,26
DNA constructs, virus expression system, and VWF enzyme-linked immunosorbent assay
The construction of NMII-A wild-type (WT) was generated by amplifying the human MYH9 gene from the cDNA of HUVECs and ligating it into the PINCO vector (Addgene) flag-tagged in the N-terminal. The S1943A and MYH9-RD mutants were generated by overlapped extension polymerase chain reaction (PCR) using NMII-A WT as template. The overexpression and shRNA-expressing viruses for HUVECs infection were prepared as before.13,24 The medium of confluent HUVECs was collected, and the relative amounts of VWF were determined by enzyme-linked immunosorbent assay (ELISA) as previously described.24 The lysates from 3 parallel control wells of HUVECs were harvested to determine the total VWF level. The level of VWF secretion is presented as a percentage of the total VWF level.
Western blotting and immunoprecipitation analysis
Confluent HUVECs were starved in serum-free M199 medium supplemented with 1% BSA for 16 hours. The cells were stimulated with forskolin (10 mmol/L), washed twice in cold phosphate-buffered saline, and lysed in RIPA buffer containing protease and phosphatase inhibitor (Thermo, A32961). The lysates were fractionated on 8% SDS–polyacrylamide gel electrophoresis by standard western blotting analysis. For immunoprecipitation, the lysates were incubated with anti-flag M2 affinity gel (Sigma, A2220) at 4°C for 16 hours, eluted by 100 mg/mL of FLAG peptide (Sigma, F3290) in tris-buffered saline, and then analyzed by western blotting.
Antibodies, immunofluorescence staining, and confocal microscope imaging
The following antibodies were used for western blotting and immunoprecipitation: rabbit polyclonal antibody to VWF (Dako, A0082), rabbit polyclonal antibody to NMII-A (Sigma, M8064), rabbit polyclonal antibody to phospho-NMII-A Ser1943 (CST, D7Z7T), rabbit polyclonal antibody to phospho-NMII-A Ser1916 (CST, MP5191), mouse monoclonal antibody to α-tubulin (Sigma, T6199), mouse monoclonal antibody to flag (Sigma, F3165), rat monoclonal antibody to flag (Sigma, SAB4200071), mouse monoclonal antibody to Gα12 (Santa Cruz, sc-515445), mouse monoclonal antibody to Gαq (Santa Cruz, sc-136181), mouse monoclonal antibody to Rab27a (Santa Cruz, sc-74586), mouse monoclonal antibodies to zyxin (Abnova, H00007791-M01), mouse monoclonal antibody to CKIIα (Santa Cruz, SC-12738), mouse monoclonal antibody to β-actin (Sigma, A5441), and HRP-conjugated secondary antibodies (GE, anti-rabbit NA9340 and anti-mouse NA9310). Confluent ECs with or without forskolin stimulation (10 mmol/L) were fixed and immunostained as previously described.24 Actin filaments were labeled with Alexa Fluor 488-conjugated phalloidin (A12379) along with secondary Alexa Fluor–conjugated donkey anti-IgG antibodies (A21206, A21424, A10042, A31573, and A37114) from Invitrogen. The images were acquired using a 40× oil-immersion lens and analyzed under a laser scanning confocal microscope (Zeiss LSM 510). Z-stacks were processed and analyzed in ImageJ.
VWF multimer gels
The medium of confluent HUVECs after forskolin stimulation (2 μL) was denatured at 60°C for 20 minutes with 10 μL of sample buffer (70 mM Tris, pH 6.8, 4 mM EDTA, 2.4% SDS, 54% urea, 0.01% bromphenol blue); subsequently, the proteins were fractionated on a 1% agarose gel. VWF multimers were detected by incubation with rabbit anti-VWF antibody followed by goat antirabbit IgG. Densitometry analysis using ImageJ software determined the ratio of high to low molecular weight of VWF multimers.27
Mass spectrometry and gene ontology enrichment analysis
The protein samples collected by immunoprecipitation were analyzed by mass spectrometry (MS). The proteins were digested by porcine trypsin (sequencing grade modified; Promega, Madison, WI) overnight at 37°C, and the tryptic peptides were extracted with 80% acetonitrile and 0.1% formic acid (FA). The samples were dried in a vacuum centrifuge concentrator at 30°C and resuspended in 10 μL 0.1% FA. Using the Easy-nLC 1200 system, 5 μL of samples was loaded at 0.3 μL/min in 0.1% FA onto a trap column and eluted across a fritless analytical resolving column (C18, Acclaim PepMap 75 μm × 15 cm nanoViper RSLC Thermo). Peptides were injected into Thermo Orbitrap Fusion Lumos for full-scan MS spectra by an Orbitrap mass analyzer (m/z range: 300-1500 Da) with the resolution set to 60 000 (full width at half maximum) at m/z 200 Da. The MS data were aligned with the Escherichia coli Swiss-Prot database using Proteome Discoverer 2.2 software. Gene ontology (GO) enrichment was performed using the GO database (DOI: 10.5281/zenodo.5080993, 2021-07-02 released) and the Panther version 16 tool.28-31 The interactomes of NMII-A WT and E1841K mutant were classified according to the GO biological process analysis.
Generation of EC/megakaryocyte-specific NMII-A E1841K mutant mice
Myh9 E1841Kflox/flox mice were generated using a targeting construct as depicted in supplemental Figure 6A by Beijing Biocytogen. Genotyping was performed using the following primers:
E1841K forward (P1) 5′-TACACATAAGGCACGTAGTGGG-3′
E1841K reverse (P2) 5′-AACAGGAAACACCCAGGCCTAACAG-3′
E1841K recombined reverse (P3) 5′-CCAGAGAAGTCATTCAGTACTACGCG-3′
Cre forward 5′-GCCTGCATTACCGGTCGATGC-3′
Cre reverse 5′-CAGGGTGTTATAAGCAATCCC-3′
WT PCR product (P1,P2) = 466 bp; mutant loxP PCR product (P1,P2) = 590 bp; mutant nonrecombined PCR product (P1,P3) = 4529 bp; mutant recombined PCR product (P1,P3) = 550 bp; Cre PCR product = 500 bp. Two probes were used to screen colonies by Southern blotting (supplemental Figure 6C). Myh9 E1841Kflox/flox mice were crossed with Tie2-Cre32 (from Professor Xiao Yang, Peking University Health Science Center, Beijing, China) or PF4-Cre mice33 (from Professor Junling Liu, Shanghai Jiao Tong University School of Medicine, Shanghai, China) to obtain endothelium- and megakaryocyte-specific NMII-A E1841K mutant mice. The mice were maintained in the Center for Experimental Animals (a facility accredited by the Association for Assessment and Accreditation of Laboratory Animal Care) at Peking University, Beijing, China, under a 12-hour light/12-hour dark cycle with free access to food and water. All procedures involving animals followed protocols approved by the Committee for Animal Research of Peking University and followed the Guide for the Care and Use of Laboratory Animals.
Measurement of plasma VWF
Ten-week-old mice were injected with epinephrine (0.5 mg/kg, intraperitoneal [IP]). Before and after 30 minutes of injection, blood samples were collected from the tail vein into tubes with 3.2% sodium citrate. Plasma VWF levels were measured using ELISA as previously described.24
Bleeding time
Ten-week-old mice were injected with epinephrine (0.5 mg/kg, IP). After 30 minutes of injection, the distal 3-mm segment of the tail was amputated to measure bleeding time according to the previously described methods.13
Thrombus formation in mesenteric vessels
Ten-week-old mice were injected with epinephrine (0.5 mg/kg, IP), and the thrombus formation happened according to the previously described methods.13 Peripheral blood was collected from the tail vein of 10-week-old mice and centrifuged at 250g for 10 minutes to obtain platelet-rich plasma. Platelets were labeled with rhodamine (2.5 mg/L). Rhodamine-labeled platelets and FITC-conjugated anti-VWF antibody (1 mg/mL) were injected into the tail vein of 10-week-old mice. After midline abdominal incision, the mesentery was exteriorized for visualizing the mesenteric veins. The mesenteric veins were applied by a filter paper (2 × 1 mm) saturated with 20% FeCl3 for 2 minutes and then monitored for 20 minutes.
Measurement of plasma FVIII
Blood samples were collected from the tail vein of 10-week-old mice into tubes with 3.2% sodium citrate. Plasma FVIII levels were measured using a mFVIII ELISA assay (Meimian, MM-0019M2) according to manufacturer's protocol. Pooled plasma from WT C57Bl/6 mice was used for the standard curve.
Activated partial thromboplastin time
Blood samples were collected from the tail vein of 10-week-old mice into tubes with 3.2% sodium citrate and centrifuged for 10 minutes at 18 000g at 4°C to obtain platelet-poor plasma. Activated partial thromboplastin time (APTT) was measured by standard assay (PSAITONG, PS0426) according to manufacturer's protocol. Platelet-poor plasma samples were mixed with APTT reagent for 3 minutes at 37°C. Subsequently, 25 mM of CaCl2 was added, and the time to clot formation was measured.
Megakaryocyte isolation and proplatelet formation
Mouse bone marrow was flushed from femurs and tibias with serum-free DMEM. The cells were cultured in DMEM supplemented with 10% fetal bovine serum, 2 mM of l-glutamine, 50 U/mL of penicillin, 50 μg/mL of streptomycin, and 50 ng/mL of thrombopoietin. Live proplatelets were visualized with the inverted fluorescence microscope after 5 days of culture.9,34
Mean platelet volume and platelet count
Whole blood was collected for mean platelet volume (MPV) and platelet count by a ProCyte Dx Hematology Analyzer.9
Platelet diameter
Wright-Giemsa–stained blood smears were visualized with the inverted fluorescence microscope. Platelet diameters were measured using ImageJ.9
Clot retraction
Whole blood was collected into tubes with 4% sodium citrate and centrifuged at 250g for 10 minutes to obtain platelet-rich plasma, which was stimulated with thrombin (10 U/mL, Sigma) in the presence of 20 mM of CaCl2 and 2 μL of erythrocytes and then incubated at 37°C for 5 hours.9,35
Statistics
All experiments were performed in at least 3 independent experiments. Data are expressed as the mean ± standard deviation (SD). ImageJ Version 1.52 and GraphPad Prism Version 8.0.2 were used for statistical analysis. Statistical differences were assessed with the unpaired two-tailed Student t test. Values of P < .05 were considered statistically significant.
Data availability
All data are included in the published article and supplemental files. All relevant materials are available from the authors upon request.
Results
Expression of MYH9-RD E1841K mutant in ECs inhibits cAMP-induced endothelial VWF secretion
Based on the relative frequency of MYH9-RD mutations,36 5 were chosen to screen for an influence on cAMP-mediated endothelial VWF secretion, 2 mutations in the globular head and 3 in the rod domain. These mutations affect the structure and function of NMII-A on multiple aspects, such as changes in conformation, ATPase activity, actin binding, and bipolar filament formation,1,11,12,37 and individuals with these mutations mostly have a bleeding tendency of variable severity. NMII-A WT and mutants were introduced into HUVECs expressing scrambled shRNA (SCR) or shRNA against NMII-A (shNMII-A) (Figure 1A), which were used for the evaluation of VWF secretion in response to forskolin, a cAMP-elevating agonist. We found that NMII-A E1841K mutant reduced cAMP-triggered VWF secretion, while the basal VWF secretion was not influenced by NMII-A mutants. Besides, unlike NMII-A WT, E1841K mutant did not rescue the defect of cAMP-induced VWF secretion in HUVECs expressing shNMII-A (Figure 1B).
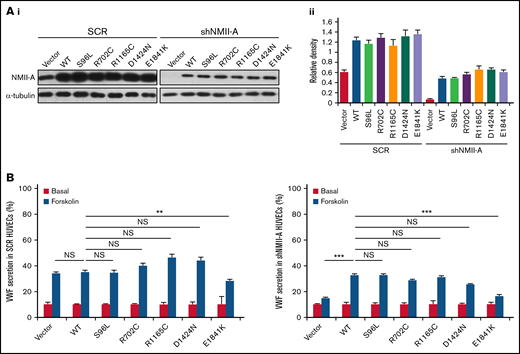
Screening of MYH9-RD mutations reveals that the expression of E1841K mutant inhibits cAMP-induced endothelial VWF secretion. (Ai) Western blots of NMII-A mutants in HUVECs expressing scrambled shRNA (SCR) or shRNA targeting NMII-A (shNMII-A) that were rescued by NMII-A WT and 5 MYH9-RD mutants. (Aii) Ratio of NMII-A to loading control α-tubulin of (Ai). (B) ELISA of VWF secreted from HUVECs in A with forskolin stimulation (n = 12; NS > 0.05; **P < .01; ***P < .001vs WT). NS, not significant.
Screening of MYH9-RD mutations reveals that the expression of E1841K mutant inhibits cAMP-induced endothelial VWF secretion. (Ai) Western blots of NMII-A mutants in HUVECs expressing scrambled shRNA (SCR) or shRNA targeting NMII-A (shNMII-A) that were rescued by NMII-A WT and 5 MYH9-RD mutants. (Aii) Ratio of NMII-A to loading control α-tubulin of (Ai). (B) ELISA of VWF secreted from HUVECs in A with forskolin stimulation (n = 12; NS > 0.05; **P < .01; ***P < .001vs WT). NS, not significant.
Subsequently, the effect of E1841K mutant on the distribution and exocytosis of WPBs was assessed by VWF immunostaining in HUVECs expressing NMII-A WT or E1841K mutant. While there was no significant difference in the number of WPBs between cells expressing NMII-A WT and E1841K mutant under basal condition, ∼70% of the WPBs in cells expressing NMII-A WT disappeared, likely as a result of exocytosis after forskolin stimulation. In contrast, a significant number of WPBs were retained in cells expressing E1841K mutant (supplemental Figure 1). These results indicate that expression of NMII-A E1841K mutant impairs cAMP-mediated WPB exocytosis and VWF secretion.
E1841K mutant influences the peripheral distribution and subsequent exocytosis of Rab27a-positive WPBs due to decreased phosphorylation of NMII-A at S1943
Phosphorylation of NMII-A at S1943 (p-S1943) is critical for WPB positioning and cAMP-induced VWF secretion.13 Therefore, we tested the effect of E1841K mutation on p-S1943 and found that p-S1943 level was significantly reduced in HUVECs expressing E1841K mutant (Figure 2A). Besides, in HUVECs expressing SCR or shNMII-A, NMII-A WT expression showed a typical myosin II pattern colocalized along stress fibers in the central region of the cells, while E1841K mutant showed a distribution with prominent localization in the periphery. This mutant also formed abnormal puncta, suggesting aberrant aggregates of E1841K mutant (supplemental Figure 2A). Interestingly, the distribution of E1841K mutant was similar to that of the nonphosphorylatable mutant of S1943 (NMII-A S1943A). Together, these data show that E1841K mutation interferes with the phosphorylation of NMII-A at S1943 and NMII-A distribution.
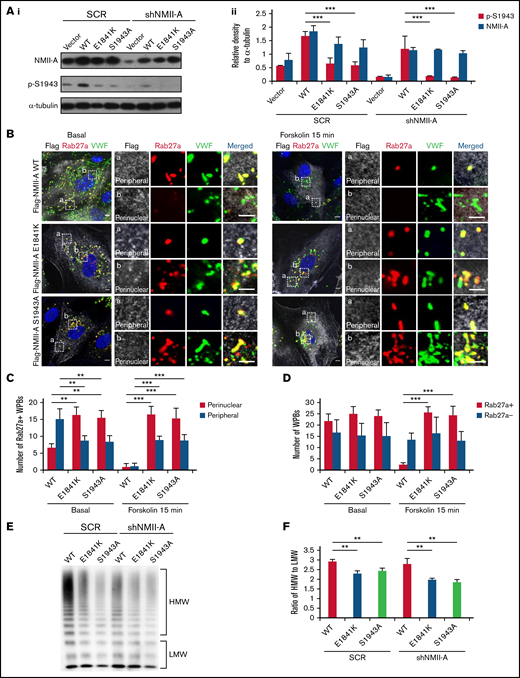
E1841K mutant influences the peripheral distribution and subsequent exocytosis of Rab27a-positive WPBs due to decreased phosphorylation of NMII-A at S1943. (Ai) Western blots of p-S1943 and total NMII-A in HUVECs expressing SCR or shNMII-A that were rescued by flag-tagged NMII-A WT, E1841K and S1943A mutant. (Aii) Ratio of p-S1943 and total NMII-A to loading control α-tubulin of Ai (***P < .001). (B) Immunostaining of VWF (green), flag (white), and Rab27a (red) in HUVECs expressing flag-tagged NMII-A WT, E1841K, and S1943A mutant. The magnified panels showed the peripheral region (a) and the perinuclear region (b). The peripheral region was defined as the region whose distance to nuclear was >10 μm. Scale bars represent 5 μm. (C) Quantitative analysis of the number of Rab27a-positive WPBs in perinuclear and peripheral regions. (D) Quantitative analysis of the number of Rab27a-negative and Rab27a-positive WPBs (n = 16; **P < .01; ***P < .001). (E) VWF multimer distribution of confluent HUVEC medium after 15 minutes of forskolin stimulation. (F) Ratios of high to low molecular weight of VWF multimers in (C) (**P < .01).
E1841K mutant influences the peripheral distribution and subsequent exocytosis of Rab27a-positive WPBs due to decreased phosphorylation of NMII-A at S1943. (Ai) Western blots of p-S1943 and total NMII-A in HUVECs expressing SCR or shNMII-A that were rescued by flag-tagged NMII-A WT, E1841K and S1943A mutant. (Aii) Ratio of p-S1943 and total NMII-A to loading control α-tubulin of Ai (***P < .001). (B) Immunostaining of VWF (green), flag (white), and Rab27a (red) in HUVECs expressing flag-tagged NMII-A WT, E1841K, and S1943A mutant. The magnified panels showed the peripheral region (a) and the perinuclear region (b). The peripheral region was defined as the region whose distance to nuclear was >10 μm. Scale bars represent 5 μm. (C) Quantitative analysis of the number of Rab27a-positive WPBs in perinuclear and peripheral regions. (D) Quantitative analysis of the number of Rab27a-negative and Rab27a-positive WPBs (n = 16; **P < .01; ***P < .001). (E) VWF multimer distribution of confluent HUVEC medium after 15 minutes of forskolin stimulation. (F) Ratios of high to low molecular weight of VWF multimers in (C) (**P < .01).
It has been previously shown that p-S1943 is required for the peripheral distribution of mature WPBs, which are Rab27a positive and largely undergo exocytosis under stimulation.13,38,,-41 We then examined whether the Rab27a-mediated cortical anchorage of WPBs is affected in E1841K mutant expressing cells. Immunostaining showed that Rab27a-positive WPBs were mostly localized in the periphery area of HUVECs expressing NMII-A WT, while they distributed in the perinuclear region in HUVECs expressing E1841K or S1943A mutant. The total number of Rab27a-positive WPBs was not significantly influenced, suggesting that E1841K or S1943A mutant does not influence the maturation of WPBs (Figure 2B-C). After forskolin stimulation, peripheral Rab27a-positive WPBs in NMII-A WT expressing HUVECs likely underwent exocytosis, while perinuclear Rab27a-positive WPBs were retained in HUVEC expressing E1841K or S1943A mutant (Figure 2D).
Because the intrinsic integrity and effective remodeling of the cortical actin network are important for WPB peripheral anchorage and exocytosis,24,41 we further investigated whether the abnormal WPB distribution was due to defective cortical actin structure in HUVECs expressing E1841K mutant. In contrast to WT-expressing ECs that exhibited strong stress fiber bundles with a cortical F-actin rim at the margins in the quiescent state, cells expressing E1841K or S1943A mutant showed weak stress fibers but strong cortical actin. After stimulation with forskolin, the actin cytoskeleton was significantly rearranged in WT ECs, while no further changes were detected in the actin distribution in ECs expressing E1841K or S1943A mutant (supplemental Figure 2B). The abnormalities of cortical actin structure and WPB distribution indicate a possible role of actin in the abnormal distribution of WPBs. In summary, E1841K mutant disrupts WPBs distribution, which is correlated with defective cortical actin structure, likely due to decreased p-S1943 level.
Besides, cAMP-induced secretion of VWF from ECs expressing NMII-A WT is composed of mainly ultra-large multimers, while the ratios of high molecular weight to low molecular weight of VWF secreted by HUVECs expressing E1841K or S1943A mutant were lower than HUVECs expressing NMII-A WT (Figure 2E-F), indicating the impaired WPB exocytosis by E1841K mutant because WPBs deliver ultra-large, highly multimeric VWF.42
E1841K mutant decreases the number of cAMP-induced formation of actin framework around WPBs due to decreased phosphorylation of NMII-A at S1916
Previously, we found that phosphorylation of NMII-A at S1916 (p-S1916) is required for cAMP-mediated formation of actin framework around WPBs and subsequent exocytosis.13 We thus examined whether E1841K mutation interferes with p-S1916 and actin framework formation. As shown in Figure 3A-C, cAMP-induced p-S1916 level was significantly reduced and the number of actin frameworks around WPBs was lower in cells expressing E1841K mutant than cells expressing NMII-A WT or the other 4 mutants. Besides, ∼70% of the WPBs in cells expressing NMII-A WT or the other 4 mutants disappeared, likely as a result of exocytosis. In contrast, a significant number of WPBs was retained in cells expressing E1841K mutant (Figure 3D). Together, these data show that E1841K mutant impairs actin frameworks formation around exocytotic WPBs due to decreased p-S1916 level and thus impairs cAMP-mediated endothelial WPB exocytosis.

E1841K mutant decreases the number of cAMP-induced formation of actin framework around WPBs due to decreased phosphorylation of NMII-A at S1916. (A) Western blotting of p-S1916 and total NMII-A in HUVECs expressing flag-tagged NMII-A WT and 5 MYH9-RD mutants stimulated with forskolin for 15 minutes. (B) Immunostaining of VWF (yellow), flag (magenta), and F-actin (cyan) in HUVECs expressing flag-tagged NMII-A WT and 5 MYH9-RD mutants. Scale bars represent 2 μm. (C) Quantitative analysis of the number of actin framework-coupled WPBs per cell. (D) The total number of WPBs per cell (n = 16; NS > 0.05; **P < .01). NS, not significant.
E1841K mutant decreases the number of cAMP-induced formation of actin framework around WPBs due to decreased phosphorylation of NMII-A at S1916. (A) Western blotting of p-S1916 and total NMII-A in HUVECs expressing flag-tagged NMII-A WT and 5 MYH9-RD mutants stimulated with forskolin for 15 minutes. (B) Immunostaining of VWF (yellow), flag (magenta), and F-actin (cyan) in HUVECs expressing flag-tagged NMII-A WT and 5 MYH9-RD mutants. Scale bars represent 2 μm. (C) Quantitative analysis of the number of actin framework-coupled WPBs per cell. (D) The total number of WPBs per cell (n = 16; NS > 0.05; **P < .01). NS, not significant.
E1841K mutation interferes with p-S1916 through interrupting interaction of NMII-A with CKIIα and zyxin
To define the specific mechanism underlying the dysfunction of E1841K mutant, we compared the interactomes of E1841K mutant with NMII-A WT to unravel the molecular basis of the mutation-induced pathology because NMII-A is known to exert its functions at least in part via interacting with other proteins.2,13 After expressing flag-tagged NMII-A WT or E1841K mutant in HUVECs, we harvested cell lysates before and after forskolin stimulation and collected protein binding to NMII-A by immunoprecipitation using anti-flag beads. Subsequently, the protein samples were subject to interactomic analysis by MS. Functional analysis of the interactomes by GO analysis showed that the interaction of E1841K mutant with the proteins responsible for regulating actin cytoskeleton or cytoskeleton-dependent intracellular vesicle transport was significantly decreased under both basal conditions and forskolin stimulation (supplemental Figure 3). Based on the difference of interactomes and immunoprecipitation, we found that E1841K mutant failed to interact with several molecules including zyxin and CKIIα (supplemental Figure 4A), which are known to be important for NMII-A function in regulation of VWF13 by promoting cAMP-induced p-S1916 (supplemental Figure 4B) and actin frameworks formation.13,24,43 In summary, E1841K mutation interferes with the interaction of NMII-A with CKIIα, which decreases cAMP-induced p-S1916, and with zyxin, impairing the formation of actin framework around exocytotic WPBs.
E1841K mutant does not affect Gα12-mediated basal secretion or thrombin-induced Gα12 and Gαq dependent VWF secretion
Besides cAMP-induced pathway, WPB exocytosis can occur in basal condition or after Ca2+-induced stimulation.44,45 To find out the effect of E1841K mutant on basal and Ca2+-induced VWF secretion, we expressed NMII-A WT and E1841K mutant in Gα12 or Gαq-depleted HUVECs because it has been shown that Gα12 mediates both basal and Ca2+-induced VWF secretion, and Gαq mediates Ca2+-induced VWF secretion from ECs under thrombin stimulation.44 However, we found that the expression of E1841K mutant did not affect Gα12-mediated basal secretion as well as thrombin-induced Gα12 and Gαq dependent VWF secretion (supplemental Figure 5A-B). Besides, there was no significant difference in the percentage of high or low molecular weight VWF multimers of basal VWF secretion from HUVECs expressing vector, NMII-A WT or E1841K mutant (supplemental Figure 5C-D), suggesting that E1841K mutant does not influence VWF packaging or processing. These results suggest that E1841K mutant does not affect Gα12-mediated basal secretion or thrombin-induced Gα12 and Gαq-dependent VWF secretion.
Generation of endothelium- and megakaryocyte-specific NMII-A E1841K mutant mice
To determine the relative contribution of E1841K in ECs and platelets to the bleeding phenotype of MYH9-RD, we generated tissue-specific knockin mice, which specifically expressed E1841K mutant in ECs (driven by Tie2-Cre) or megakaryocytes (driven by PF4-Cre), using the Cre-loxP recombination strategy as shown in supplemental Figure 6A. The last exon 36-41 of Myh9 was replaced through homologous recombination by a targeting cassette spanning WT exon 36-41 flanked by loxp sites and the following exon 36-41 duplication harboring the E1841K mutation. In tissues expressing cre recombinase, the WT exon 36-41 of Myh9 in the knockin allele was removed via Cre/loxP recombination. PCR genotyping primers P1 and P3 (supplemental Figure 6A) were used to verify the recombination of genomic DNA isolated from ECs/megakaryocytes of cre-, Tie2-cre and PF4-cre/E1841Kflox/flox mice. Elimination of the fragment between the 2 loxP sites by recombination resulted in a smaller PCR amplicon (from 4529 bp to 550 bp), which was verified in ECs/megakaryocytes from Tie2-cre and PF4-cre/E1841Kflox/flox mice respectively (supplemental Figure 6B). The targeting cassette introduced ScaI and XmnI restriction sites whose digestions resulted in DNA fragments of different lengths for the WT and knockin alleles. Southern blot results showed DNA fragments in the 13.8-kb band by a 5′ probe and the 8.6-kb band by a 3′ probe representing the WT allele, while the 9.5-kb band by a 5′ probe and the 10.9-kb band by a 3′ probe represented the knockin allele (supplemental Figure 6C). The protein expression levels of E1841K mutant in ECs or megakaryocytes isolated from Tie2-cre and PF4-cre/E1841Kflox/flox mice were similar to those in control cells from cre-/E1841Kflox/flox mice (supplemental Figure 6D), indicating that introduction of the E1841K mutation did not affect NMII-A expression or protein stability. The E1841K point mutation (c. 5521 G > A) was further confirmed by sequencing the cDNA of ECs and megakaryocytes isolated from Tie2-cre and PF4-cre/E1841Kflox/flox mice, respectively (supplemental Figure 6E). Collectively, these results confirm the successful generations of EC/megakaryocyte-specific E1841K mutant mice.
Endothelium-specific E1841K mutant hinders cAMP-mediated VWF release, prolongs bleeding time, and impairs thrombosis in mice
Then, we analyzed the effect of E1841K mutant in ECs on bleeding phenotype of MYH9-RD by measuring the plasma VWF level and bleeding time of these tissue-specific knock-in mice. While levels and multimer patterns of basal plasma VWF were comparable in cre-, PF4-cre and Tie2-cre/E1841Kflox/flox mice, cAMP-induced VWF release after epinephrine stimulation was significantly suppressed in Tie2-cre/E1841Kflox/flox mice, while epinephrine-induced VWF release in PF4-cre/E1841Kflox/flox mice was similar to controls (Figure 4A,C-D). Consistently, the bleeding times of the mouse tail vein in PF4-cre and Tie2-cre/E1841Kflox/flox mice were longer than the controls. After epinephrine stimulation, PF4-cre/E1841Kflox/flox and Tie2-cre/E1841Kflox/flox mice exhibited prolonged bleeding times when compared with the cre-/E1841Kflox/flox mice (Figure 4B). Heterozygous mutant mice also showed significant defect in hemostasis, although less severe than homozygous mutant mice. Similarly, after epinephrine stimulation, compared with control mice, the FeCl3-induced thrombus formation of mesenteric vessels in Tie2-cre or PF4-cre/E1841Kflox/flox mice was significantly impaired (Figure 4E-F). However, the expression of E1841K mutant in ECs or megakaryocytes did not affect plasma FVIII levels or other clotting factor activities measured by APTT (supplemental Figure 7A-B). These data demonstrate that endothelium-specific E1841K mutant disrupts cAMP-triggered endothelial VWF secretion and inhibits normal hemostasis, which may contribute to the bleeding phenotype of MYH9-RD.
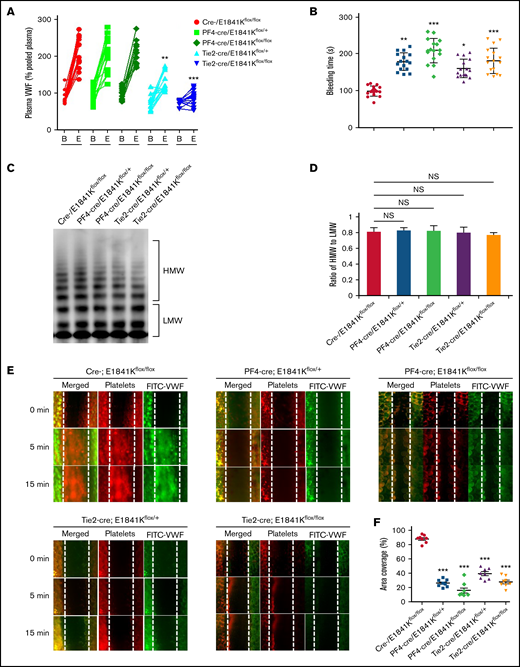
Endothelium-specific E1841K mutant hinders cAMP-mediated VWF release, prolongs bleeding time, and impairs thrombosis in mice. (A) Normalized levels of plasma VWF from cre-/E1841Kflox/flox (red), PF4-cre/E1841Kflox/+ (light green), PF4-cre/E1841Kflox/flox (dark green), Tie2-cre/E1841Kflox/+ (light blue), and Tie2-cre/E1841Kflox/flox (dark blue) (n = 14) mice before (B) and after epinephrine stimulation (E). (B) Bleeding times of cre-/E1841Kflox/flox (red), PF4-cre/E1841Kflox/+ (light green), PF4-cre/E1841Kflox/flox (dark green), Tie2-cre/E1841Kflox/+ (light blue), and Tie2-cre/E1841Kflox/flox (dark blue) (n = 16) mice after epinephrine stimulation (*P < .05; **P < .01; ***P < .001 vs cre-/E1841Kflox/flox mice). (C) VWF multimer distribution of basal mice plasma. (D) Ratios of high to low molecular weight of VWF multimers in C (NS > 0.05). (E) FeCl3-induced thrombus formation in the mesenteric vessels of cre-/E1841Kflox/flox, PF4-cre/E1841Kflox/+, PF4-cre/E1841Kflox/flox, Tie2-cre/E1841Kflox/+, and Tie2-cre/E1841Kflox/flox (n = 8) mice. The thrombus is indicated with Rhodamine-labeled platelets and FITC-conjugated anti-VWF antibody. (F) The coverage of the area by thrombus in (E) at 15 minutes (*P < .05; ***P < .001).
Endothelium-specific E1841K mutant hinders cAMP-mediated VWF release, prolongs bleeding time, and impairs thrombosis in mice. (A) Normalized levels of plasma VWF from cre-/E1841Kflox/flox (red), PF4-cre/E1841Kflox/+ (light green), PF4-cre/E1841Kflox/flox (dark green), Tie2-cre/E1841Kflox/+ (light blue), and Tie2-cre/E1841Kflox/flox (dark blue) (n = 14) mice before (B) and after epinephrine stimulation (E). (B) Bleeding times of cre-/E1841Kflox/flox (red), PF4-cre/E1841Kflox/+ (light green), PF4-cre/E1841Kflox/flox (dark green), Tie2-cre/E1841Kflox/+ (light blue), and Tie2-cre/E1841Kflox/flox (dark blue) (n = 16) mice after epinephrine stimulation (*P < .05; **P < .01; ***P < .001 vs cre-/E1841Kflox/flox mice). (C) VWF multimer distribution of basal mice plasma. (D) Ratios of high to low molecular weight of VWF multimers in C (NS > 0.05). (E) FeCl3-induced thrombus formation in the mesenteric vessels of cre-/E1841Kflox/flox, PF4-cre/E1841Kflox/+, PF4-cre/E1841Kflox/flox, Tie2-cre/E1841Kflox/+, and Tie2-cre/E1841Kflox/flox (n = 8) mice. The thrombus is indicated with Rhodamine-labeled platelets and FITC-conjugated anti-VWF antibody. (F) The coverage of the area by thrombus in (E) at 15 minutes (*P < .05; ***P < .001).
Abnormal WPB distribution in primary ECs derived from endothelium-specific E1841K mutant mice
To confirm the effect of E1841K mutant on cAMP-triggered endothelial VWF secretion in EC-specific Myh9 E1841K mutant mice, we isolated BECs from WT and mutant mice. ECs derived from endothelium-specific E1841K mutant mice displayed the distribution of WPBs in perinuclear region, accompanied by few stress fibers with strong cortical actin (Figure 5A-C). In contrast, BECs from megakaryocyte-specific E1841K mutant mice exhibited a phenotype similar to controls. Together, the data from the isolated ECs from endothelium-specific E1841K mutant mice indicate that endothelial E1841K mutant disrupts normal peripheral WPB distribution accompanied by defective cortical actin structure.
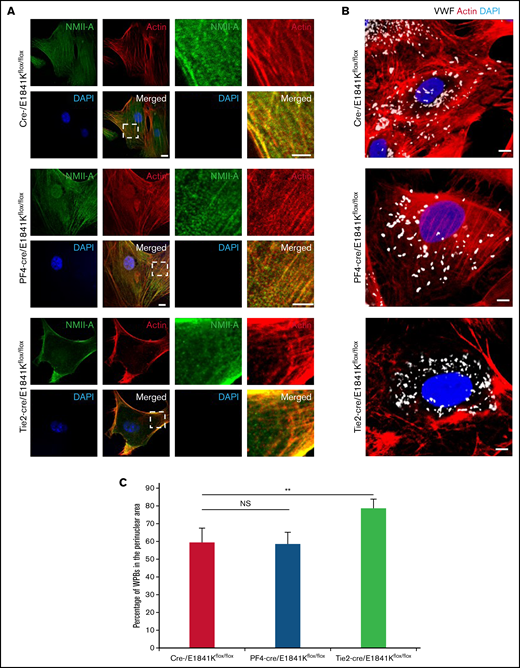
Abnormal WPB distribution in primary ECs derived from endothelium-specific E1841K mutant mice. (A) Immunostaining of NMII-A (green) and actin (red) in BECs from cre-, PF4-cre, and Tie2-cre/E1841Kflox/flox mice. Scale bars represent 5 μm. (B) Immunostaining of VWF (white) and actin (red) in these BECs. (C) Percentage of WPBs in the perinuclear area (n = 16; NS > 0.05; **P < .01). NS, not significant.
Abnormal WPB distribution in primary ECs derived from endothelium-specific E1841K mutant mice. (A) Immunostaining of NMII-A (green) and actin (red) in BECs from cre-, PF4-cre, and Tie2-cre/E1841Kflox/flox mice. Scale bars represent 5 μm. (B) Immunostaining of VWF (white) and actin (red) in these BECs. (C) Percentage of WPBs in the perinuclear area (n = 16; NS > 0.05; **P < .01). NS, not significant.
Defective cAMP-induced VWF secretion is associated with decreased number of actin framework formation in primary ECs derived from endothelium-specific E1841K mutant mice
Consistent with the earlier findings in human ECs, the cAMP-triggered VWF secretion was abolished in BECs from endothelium-specific E1841K mutant mice (Figure 6A) due to the low level of p-S1916 and p-S1943 and decreased number of actin framework formation around exocytotic WPBs in BECs from endothelium-specific E1841K mutant mice (Figure 6B-E). In contrast, BECs from megakaryocyte-specific E1841K mutant mice exhibited a phenotype similar to controls. Together, the data of the isolated ECs from endothelium-specific E1841K mutant mice indicate that endothelial E1841K mutant disrupts mouse hemostasis likely via disrupting WPB distribution and actin frameworks formation around WPBs, which hinders cAMP-mediated VWF secretion.
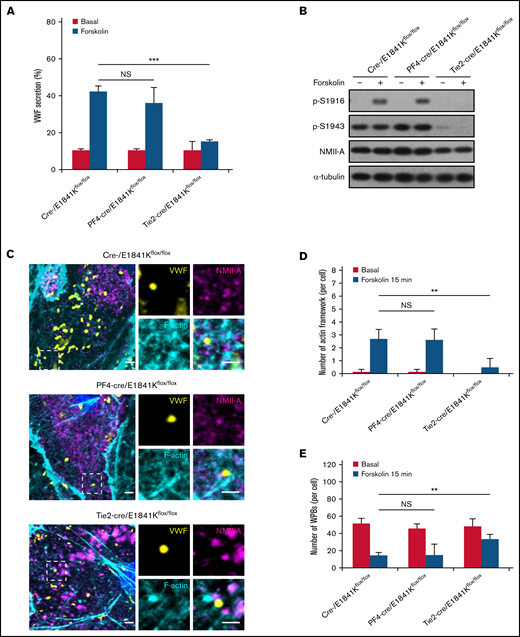
Defective cAMP-induced VWF secretion is associated with decreased number of actin framework formation in primary ECs derived from endothelium-specific E1841K mutant mice. (A) VWF secretion in BECs from cre-, PF4-cre, and Tie2-cre/E1841Kflox/flox mice with forskolin stimulation (NS > 0.05; ***P < .001). (B) Western blotting of p-S1916, p-S1943, and total NMII-A in BECs from cre-, PF4-cre, and Tie2-cre/E1841Kflox/flox mice stimulated with forskolin for 15 minutes. (C) Immunostaining of VWF (yellow), NMII-A (magenta), and F-actin (cyan) in BECs from cre-, PF4-cre, and Tie2-cre/E1841Kflox/flox mice. Scale bars represent 2 μm. (D) Quantitative analysis of the number of actin framework-coupled WPBs per cell. (E) The total number of WPBs per cell (n = 16; NS > 0.05; **P < .01). NS, not significant.
Defective cAMP-induced VWF secretion is associated with decreased number of actin framework formation in primary ECs derived from endothelium-specific E1841K mutant mice. (A) VWF secretion in BECs from cre-, PF4-cre, and Tie2-cre/E1841Kflox/flox mice with forskolin stimulation (NS > 0.05; ***P < .001). (B) Western blotting of p-S1916, p-S1943, and total NMII-A in BECs from cre-, PF4-cre, and Tie2-cre/E1841Kflox/flox mice stimulated with forskolin for 15 minutes. (C) Immunostaining of VWF (yellow), NMII-A (magenta), and F-actin (cyan) in BECs from cre-, PF4-cre, and Tie2-cre/E1841Kflox/flox mice. Scale bars represent 2 μm. (D) Quantitative analysis of the number of actin framework-coupled WPBs per cell. (E) The total number of WPBs per cell (n = 16; NS > 0.05; **P < .01). NS, not significant.
Endothelium-specific E1841K mutant mice show normal platelets while megakaryocyte-specific E1841K mutant mice show platelet abnormalities characteristic of MYH9-RD
Macrothrombocytopenia is a common clinical manifestation in both patients with MYH9-RD and mouse models.4,9 To analyze the functions of megakaryocyte and platelet in E1841K mutant mice, megakaryocytes were cultured from bone marrow with thrombopoietin stimulation for 5 days. Live-cell images showed that proplatelet formation in megakaryocytes from Tie2-cre/E1841Kflox/flox mice was similar to that from controls, whereas megakaryocytes from PF4-cre/E1841Kflox/flox mice exhibited much reduced proplatelet formation (Figure 7A). Wright-Giemsa staining of blood smears showed normal platelets from Tie2-cre/E1841Kflox/flox mice and many abnormally large platelets from PF4-cre/E1841Kflox/flox mice (Figure 7B). The MPVs derived from Tie2-cre/E1841Kflox/flox mice were the same as controls, but those from PF4-cre/E1841Kflox/flox mice were ∼1.5 times larger (Figure 7C). To confirm the MPV results, the platelet diameters were also measured from the blood smears: those from Tie2-cre/E1841Kflox/flox mice were comparable to controls, whereas those from PF4-cre/E1841Kflox/flox mice were ∼1.3 times larger (Figure 7D). Besides, Tie2-cre/E1841Kflox/flox mice had the same number of platelets as controls, while PF4-cre/E1841Kflox/flox mice had markedly lower platelet counts, only ∼50% of that from cre- or Tie2-cre/E1841Kflox/flox mice (Figure 7E). Platelet function was normal in Tie2-cre/E1841Kflox/flox mice but impaired in PF4-cre/E1841Kflox/flox mice characterized by a defect in clot retraction, a platelet-dependent contractile process (Figure 7F). Heterozygous PF4-cre/E1841Kflox/+ mice also showed platelet abnormalities, although less severe than the phenotypes of homozygous PF4-cre/E1841Kflox/flox mice. Collectively, these results show that megakaryocyte and platelet functions are normal in endothelium-specific E1841K mutant mice, while the megakaryocyte-specific E1841K mutant results in disrupted proplatelet formation, and macrothrombocytopenia with impaired platelet function.
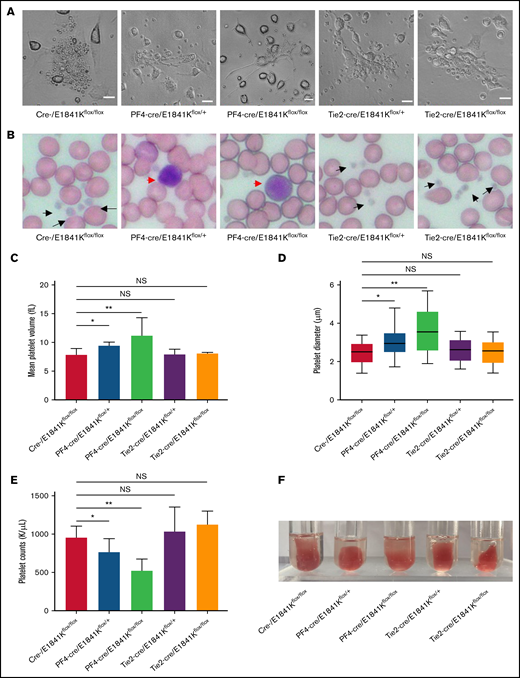
Endothelium-specific E1841K mutant mice show normal platelets while megakaryocyte-specific E1841K mutant mice show platelet abnormalities characteristic of MYH9-RD. (A) Live-cell images of proplatelet formation from isolated megakaryocytes. Scale bars represent 20 μm. (B) Wright-Giemsa–stained blood smears from cre-, Tie2-cre/E1841Kflox/+, and Tie2-cre/E1841Kflox/flox mice show normal size platelets (black arrows), while blood smears from PF4-cre/E1841Kflox/+ and PF4-cre/E1841Kflox/flox mice show abnormally large platelets (red arrow). (C) Mean platelet volumes of circulating platelets. Results represent mean ± SD. MPVs of cre-/E1841Kflox/flox: 7.86 ± 1.09 fL (n = 16); PF4-cre/E1841Kflox/+: 9.44 ± 0.58 fL (n = 16); PF4-cre/E1841Kflox/flox: 11.18 ± 3.14 fL (n = 16); Tie2-cre/E1841Kflox/+: 7.88 ± 0.94 fL (n = 16); and Tie2-cre/E1841Kflox/flox: 8.04 ± 0.22 fL (n = 16) (NS > 0.05; *P < .05; **P < .01). (D) Platelet diameters measured from peripheral blood smears. Platelet diameters (mean ± SD) of cre-/E1841Kflox/flox: 2.45 ± 0.58 μm (n = 150); PF4-cre/E1841Kflox/+: 3.02 ± 0.67 μm (n = 150); PF4-cre/E1841Kflox/flox: 3.66 ± 1.14 μm (n = 150); Tie2-cre/E1841Kflox/+: 2.59 ± 0.57 μm (n = 150); and Tie2-cre/E1841Kflox/flox: 2.51 ± 0.59 μm (n = 150) (NS > 0.05; *P < .05; **P < .01). (E) Circulating platelet counts from peripheral blood. Results represent mean ± SD. Circulating platelet counts of cre-/E1841Kflox/flox: 954.75 ± 152.63 K/μL (n = 16); PF4-cre/E1841Kflox/+: 764.12 ± 175.43 K/μL (n = 16); PF4-cre/E1841Kflox/flox: 522.19 ± 154.08 K/μL (n = 16); Tie2-cre/E1841Kflox/+: 1029.56 ± 322.01 K/μL (n = 16); and Tie2-cre/E1841Kflox/flox: 1124.44 ± 176.18 K/μL (n = 16) (NS > 0.05; *P < .05; **P < .01). (F) Clot retraction test 5 hours after stimulation (n = 5 per genotype). NS, not significant.
Endothelium-specific E1841K mutant mice show normal platelets while megakaryocyte-specific E1841K mutant mice show platelet abnormalities characteristic of MYH9-RD. (A) Live-cell images of proplatelet formation from isolated megakaryocytes. Scale bars represent 20 μm. (B) Wright-Giemsa–stained blood smears from cre-, Tie2-cre/E1841Kflox/+, and Tie2-cre/E1841Kflox/flox mice show normal size platelets (black arrows), while blood smears from PF4-cre/E1841Kflox/+ and PF4-cre/E1841Kflox/flox mice show abnormally large platelets (red arrow). (C) Mean platelet volumes of circulating platelets. Results represent mean ± SD. MPVs of cre-/E1841Kflox/flox: 7.86 ± 1.09 fL (n = 16); PF4-cre/E1841Kflox/+: 9.44 ± 0.58 fL (n = 16); PF4-cre/E1841Kflox/flox: 11.18 ± 3.14 fL (n = 16); Tie2-cre/E1841Kflox/+: 7.88 ± 0.94 fL (n = 16); and Tie2-cre/E1841Kflox/flox: 8.04 ± 0.22 fL (n = 16) (NS > 0.05; *P < .05; **P < .01). (D) Platelet diameters measured from peripheral blood smears. Platelet diameters (mean ± SD) of cre-/E1841Kflox/flox: 2.45 ± 0.58 μm (n = 150); PF4-cre/E1841Kflox/+: 3.02 ± 0.67 μm (n = 150); PF4-cre/E1841Kflox/flox: 3.66 ± 1.14 μm (n = 150); Tie2-cre/E1841Kflox/+: 2.59 ± 0.57 μm (n = 150); and Tie2-cre/E1841Kflox/flox: 2.51 ± 0.59 μm (n = 150) (NS > 0.05; *P < .05; **P < .01). (E) Circulating platelet counts from peripheral blood. Results represent mean ± SD. Circulating platelet counts of cre-/E1841Kflox/flox: 954.75 ± 152.63 K/μL (n = 16); PF4-cre/E1841Kflox/+: 764.12 ± 175.43 K/μL (n = 16); PF4-cre/E1841Kflox/flox: 522.19 ± 154.08 K/μL (n = 16); Tie2-cre/E1841Kflox/+: 1029.56 ± 322.01 K/μL (n = 16); and Tie2-cre/E1841Kflox/flox: 1124.44 ± 176.18 K/μL (n = 16) (NS > 0.05; *P < .05; **P < .01). (F) Clot retraction test 5 hours after stimulation (n = 5 per genotype). NS, not significant.
Discussion
As one of the most frequent forms of inherited thrombocytopenia, the bleeding tendency in MYH9-RD has been attributed to platelet abnormalities.36 In this study, we demonstrate that E1841K, one of most common NMII-A mutations, hinders cAMP-triggered secretion of VWF from HUVECs and mouse ECs. Using a tissue-specific knockin mouse models, we show that endothelium-specific E1841K mutant mice exhibit impaired cAMP-induced VWF release. In addition, they have a prolonged bleeding time and impaired thrombosis with normal platelets, which are similar to megakaryocyte-specific mutant mice after epinephrine stimulation (Figure 4). Megakaryocyte-specific E1841K mutant mice exhibit macrothrombocytopenia and a prolonged bleeding time with normal VWF release. Thus, these data demonstrate that the impaired VWF secretion due to the expression of E1841K mutant in ECs may contribute to the bleeding phenotype of MYH9-RD that has been attributed to thrombocytopenia so far.
Using SCR/shNMII-A-expressing HUVECs rescued by NMII-A mutants, the E1841K was found, among 5 common MYH9-RD mutations, to inhibit cAMP-mediated VWF secretion (Figure 1). Given the roles of p-S1943 of NMII-A in regulation of peripheral distribution under quiescent condition, E1841K mutant impairs the peripheral distribution of Rab27a-positive WPBs likely due to decreased p-S1943 level and abnormal cortical actin remodeling (supplemental Figure 2), which may also affect subsequent cAMP-mediated Rab27a-positive WPB exocytosis (Figure 2D).13,38 -41 On the other hand, E1841K mutant-expressing ECs displayed impaired cAMP-induced actin framework formation around WPBs, which facilitates fusion and secretion (Figure 3B-D).24 In a previous study,13 we found that p-S1916 was essential for cAMP-induced formation of actin frameworks around exocytotic WPBs and VWF secretion, dependent on the interaction of NMII-A with zyxin and CKIIα. Interestingly, E1841K mutant also displayed a decreased p-S1916 level (Figure 3A) likely due to impaired interaction with zyxin and CKIIα (supplemental Figure 4). Although consistent with our previous findings, it is still unknown how the mutation of E1841, nonphosphorylated and far away from S1916, affects the interaction of NMII-A with zyxin and CKIIα and is worth further investigation. Together, these results suggest that MYH9-RD E1841K mutation may compromise endothelial NMII-A functions by interfering with its interactomes and phosphorylation, which leads to abnormal WPBs distribution and impairs actin frameworks formation around exocytotic WPBs, which consequently inhibits WPB exocytosis.
To dissect the contribution of megakaryocyte vs endothelial E1841K mutant to the bleeding phenotype, we have generated endothelium- or megakaryocyte-specific E1841K mutant mice (supplemental Figure 6). Interestingly, compared with control mice, endothelium-specific E1841K mutant mice, although they had normal platelets, exhibited a defect in hemostasis similar to megakaryocyte-specific mutant mice after epinephrine stimulation (Figure 4).
It is somewhat striking to observe only a minor reduction in basal plasma levels of VWF in the mutant endothelium-specific knockin mouse, and it primarily affected cAMP-dependent VWF secretion. In a recent study, we observed that basal plasma levels of VWF were almost normal in endothelium-specific knockout mice of NMII-A.13 Along this line, downregulation of NMII-A or expression of E1841K mutant has no effect on basal VWF secretion in HUVECs (Figure 1). A previous study showed that Gα12 mediates basal secretion via directly interacting with αSNAP, 1 component in core secretory machinery facilitating WPB exocytosis at the plasma membrane.44 However, we see little evidence in literature that NMII-A directly interacts with any component in core secretory machinery. In fact, E1841K mutant expression did not affect Gα12-mediated basal secretion (supplemental Figure 5), further suggesting that NMII-A is dispensable for basal VWF secretion in ECs. On the other hand, we previously showed an essential role of zyxin, the interacting protein of NMII-A, in regulated VWF secretion by cAMP, but not Ca2+, agonists.13 Consistently, in the present study, we showed that E1841K mutant expression affected cAMP-dependent VWF secretion without apparent effect on thrombin-induced VWF secretion (supplemental Figure 5). After WPB fusion with the plasma membrane, WPBs undergoing full fusion can exploit the contractile properties of actomyosin rings to forcibly release high molecular weight VWF, dependent on NMII activity.24,46 The cAMP-induced phosphorylation of NMII-A at S1916 is required for its interaction with zyxin and actomyosin ring formation, which is impaired in E1841K mutant expressing cells (Figure 3; supplemental Figure 4). Notably, as discussed in a recent review46 and mentioned in a recent article,47 during WPB exocytosis, cAMP-triggered actin framework formation is quite different from Ca2+ pathway-mediated cortical actin remodeling in terms of the role, timing, and percentage of actin coat assembly. Thus, it is not surprising that E1841K mutant expression had no obvious effect on thrombin-induced VWF secretion although Gα12 and/or Gαq mediates thrombin-induced VWF secretion through p115 RhoGEF and RhoA signals that are closely related to cortical actin remodeling.44
The tissue-specific E1841K mutant mice provide the chance to investigate as-yet unrecognized endothelial vs megakaryocytic defects due to the E1841K mutation. In addition, they could be used for studying new therapies for patients with MYH9-RD. Accumulating evidence has shown cross-talk between ECs and other tissue cells in organs such as kidney48,49 and liver50,51 that are rich in blood vessels. Thus, our study further encourages functional evaluation of endothelial MYH9 mutations that may contribute to MYH9-RD–associated conditions in kidney and liver.
In summary, our work suggests that the expression of E1841K mutant in ECs impairs cAMP-induced VWF secretion, which may contribute to the bleeding phenotype of MYH9-RD. Our findings shed new light on the pathogenesis of MYH9-RD and call for evaluation of the role of endothelial VWF in patients with MYH9-RD.
Acknowledgments
The authors thank Iain Bruce for his critical reading of the manuscript. The mass spectrometry analysis was performed at the National Center for Protein Sciences at Peking University with the help of L. Dong. The Tie2-cre mice were kindly gifted from Xiao Yang at State Key Laboratory of Proteomics, Beijing Proteome Research Center, and National Center for Protein Sciences (Beijing).
This study was supported by research grants from the National Natural Science Foundation of China (81930011, 91853134, 31771263, and 31970777), the National Key R&D Program of China (2019YFA0801603), and the Peking University Clinical Scientist Program (BMU2019LCKXJ001) from the Fundamental Research Funds for the Central Universities.
Authorship
Contribution: J. Luo and J.Z. designed the experiments and wrote the manuscript; Y.C. carried out the experiments, analyzed data, and wrote the manuscript; Y.S., Y.D., C.D., X.K., and Y.H. performed the experiments; G.W. designed the experiments and read the manuscript; and J. Liu and S.J. contributed vital analytical tools.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Jincai Luo, Institute of Molecular Medicine/College of Future Technology, Peking University, Beijing 100871, China; e-mail: jincailuo@pku.edu.cn; and Jingjing Zhang, Affiliated Hospital of Guangdong Medical University & Key Laboratory of Zebrafish Model for Development and Disease of Guangdong Medical University, Zhanjiang 524001, China; e-mail: jingjing.zhang@live.com.

