


TO THE EDITOR:
Toxic activation of immune cells can lead to life-threatening conditions, all previously named hemophagocytic lymphohistiocytosis (HLH). Increased knowledge of the etiology and various underlying pathologies deems new nomenclature necessary.1 HLH and HLH mimicries are life-threatening diseases characterized by uncontrolled immune hyperactivation. They are caused by overactivation of lymphocytes and macrophages producing large amounts of interferon-γ, interleukin-1β (IL-1β), and IL-18. Different underlying etiologies, including genetic defects, autoimmunity, infections, and malignancies are increasingly recognized, adding to the debate about classification.
Primary HLH is an inherited inborn error of immunity (IEI), whereas all other forms are acquired as a complication of various underlying conditions, including infection, malignancy, and autoimmunity and of allogeneic hematopoietic stem cell transplantation (allo-HSCT).2-4 Irrespective of the etiology, HLH typically presents with persistent high fever, hepatosplenomegaly, and cytopenias.2,5 Diagnosis is often based on the criteria of the HLH-2004 protocol, in which at least 5 of 8 criteria have to be fulfilled (supplemental Table 1).5 When left untreated, HLH is almost always fatal, but even treated HLH has a high mortality. Therefore, timely diagnosis and treatment are crucial.3,4,6 -8
Primary HLH is caused by a limited number of gene defects and may present at any age, but onset is mostly at a young age.7 HLH has also been reported to be a complication in one-third of the patients with an extremely rare IEI characterized by subcutaneous panniculitislike T-cell lymphoma (SPTCL). SPTCL is a rare T-cell lymphoma in which subcutaneous adipose tissue is infiltrated by αβ CD8+ T-cells or γδ T-cells.9,10 A small number of studies in patients with SPTCL have reported variants in HAVCR2, encoding T-cell immunoglobulin and mucin domain–containing protein 3 (TIM-3).11-14
TIM-3 is a member of the TIM-family of immunoregulatory proteins expressed by various immune cells and has been associated with regulation of immune responses in autoimmunity and cancer.15 Variants described in these studies result in reduced or absent TIM-3 expression, persistent immune activation and increased production of tumor necrosis factor-α and IL-1β,11,12 which could lead to a toxic immune activation cascade, as was seen in this exceptional case of a patient presenting with features reminiscent of HLH and hyperinflammation but complete lack of cytopenia. In the presence of undetectable natural killer (NK)-cell function, the initial routine HLH gene panel was found to be negative. More extensive genetic analysis revealed 2 heterozygous variants in HAVCR2, leading to absent TIM-3 expression.
A previously healthy 30-year-old man presented with persistent fever, obstipation, and 12-kg weight loss, followed by episodic fever, ascites, and extensive peripheral edema. Laboratory analysis showed normal blood cell counts apart from lymphopenia and increased liver function levels, with strongly elevated plasma ferritin levels, soluble CD25 (sIL2Rα), lactate dehydrogenase, and D-dimers. Infections, including Epstein-Barr virus, cytomegalovirus, adenovirus, and SARS-CoV-2, were excluded (supplemental Table 2; Figure 1A). Screening for autoantibodies, including antinuclear antibodies, anti-double strand DNA, anti-CCP, anti-MPO, and anti-PR3, was negative. Also, the patient did not have a medical history of autoimmune disease. Fluorodeoxyglucose-positron emission tomography (FDG-PET) revealed hepatosplenomegaly and extensive mesenteric and mesorectal fat infiltration (Figure 1A). Omental biopsies showed infiltration of CD4+ and CD8+ lymphocytes between mature adipocytes (Figure 1B) with histopathological signs of fat necrosis suggestive of panniculitis. Hemophagocytosis was noted in bone marrow and liver biopsies (Figure 1C). T-cell clonality assays were performed in omental tissue, as well as in liver and bone marrow biopsy specimens and showed polyclonal rearrangement, excluding the diagnosis of (α/β) T-cell lymphoma.
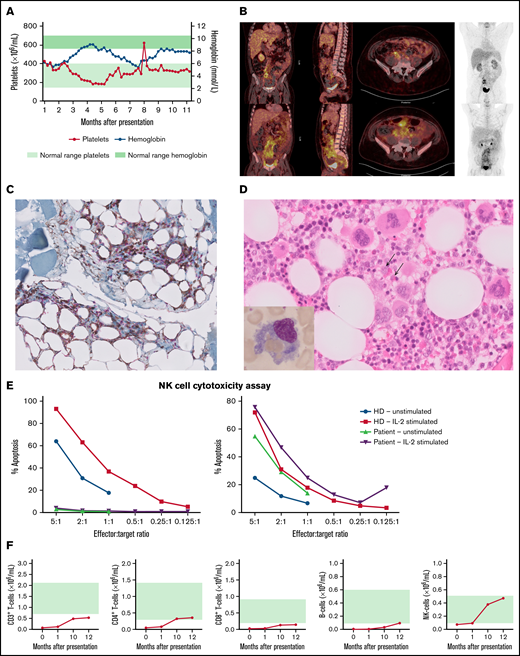
Clinical and immunological findings. (A) Absolute platelet numbers and hemoglobin levels over time (from acute phase until the present time). (B) FDG-PET showing hepatosplenomegaly and extensive mesenteric fat infiltration. (C) CD4/CD8 double-stained omental biopsy specimen (original magnification ×40), with CD4+ lymphocytes stained brown and CD8+ lymphocytes stained red, infiltrating between the mature adipocytes. (D) Hematoxylin and eosin–stained bone marrow biopsy specimen (original magnification ×40), showing trilineage hematopoiesis and scattered histiocytic cells with packed erythrocytes in the cytoplasm. Inset in the left bottom corner is Giemsa-stained bone marrow aspirate (original magnification ×100), highlighting a hemophagocytic histiocyte. (E) NK-cell function before (left) and after (right) treatment. (F) Absolute B-cell, T-cell, and NK-cell counts over time (from acute phase until the present time). Green shading indicates age-dependent normal values.
Clinical and immunological findings. (A) Absolute platelet numbers and hemoglobin levels over time (from acute phase until the present time). (B) FDG-PET showing hepatosplenomegaly and extensive mesenteric fat infiltration. (C) CD4/CD8 double-stained omental biopsy specimen (original magnification ×40), with CD4+ lymphocytes stained brown and CD8+ lymphocytes stained red, infiltrating between the mature adipocytes. (D) Hematoxylin and eosin–stained bone marrow biopsy specimen (original magnification ×40), showing trilineage hematopoiesis and scattered histiocytic cells with packed erythrocytes in the cytoplasm. Inset in the left bottom corner is Giemsa-stained bone marrow aspirate (original magnification ×100), highlighting a hemophagocytic histiocyte. (E) NK-cell function before (left) and after (right) treatment. (F) Absolute B-cell, T-cell, and NK-cell counts over time (from acute phase until the present time). Green shading indicates age-dependent normal values.
The NK-cell cytotoxicity assay showed no detectable NK-cell function (Figure 1D). The patient met 6 of 8 criteria for diagnosis of HLH, according to the HLH-2004 protocol.5 His parents were unrelated, and the family history was negative for malignancies or inflammatory disease. The primary HLH gene panel of 8 genes showed no variants.
In the absence of any underlying condition including malignancy, the immune activation syndrome was treated with etoposide and dexamethasone according to the HLH-94 protocol16 for 10 weeks. During this treatment, more extensive analysis of 419 IEI genes17 revealed compound heterozygosity for 2 variants in HAVCR2 encoding TIM-3. After careful literature review, treatment with only immunosuppressive agents (cyclosporine and prednisolone) was continued instead of extended chemotherapy combined with allo-HSCT. To date (1 year later), after cautious withdrawal of prednisolone, the patient is in complete remission with low-dose cyclosporine as the only immune suppression.
Peripheral blood samples were collected after obtaining informed consent. Whole-exome sequencing and extensive immunophenotyping were performed as described previously.18,19 Detailed methods are given in the supplemental Materials.
Whole-exome sequencing revealed 2 variants, c.291A>G (p.Ile97Met) and c.302C>T (p.Thr101Ile), in HAVCR2 encoding TIM-3 (supplemental Figure 1). Both variants have an autosomal recessive inheritance pattern and have been described in patients with HLH-SPTCL.11-14 Although the results of FDG-PET were suspect, no T-cell lymphoma was found. Our patient suffered from a prolonged febrile inflammatory disease with biomarkers compatible with toxic activation of immune cells and hemophagocytosis by histology, in the absence of cytopenia. The severe T- and B-cell lymphopenia (Figure 1E) was explained by hepatosplenomegaly and lymphocyte infiltration into the mesenteric tissue. The macrophage dysregulation is considered the primary cause of the patient’s clinical picture which is best described as an HLH-mimic disease or dysregulated immune activation or proliferation to indicate a group of gene defects with putative or proven roles in dysregulated immune activation or proliferation.1,20
Because of the atypical disease presentation, extensive immunophenotyping was performed to gain insight into the effect of the genetic variants. B-cell phenotyping showed a high percentage of switched memory B-cells and a corresponding low percentage of naive B-cells (supplemental Figure 2A). T-cell phenotyping showed a high percentage of CD4+CD45RA− memory T-cells and a low percentage of naive CD4+CD45RA+ T-cells (supplemental Figure 2B). CD8+ T-cell subsets were comparable to those of healthy donors (HDs), as was the percentage of CD4+CD25+FoxP3+ regulatory T-cells (supplemental Figure 2C and data not shown). Initially, the patient also had an increased percentage of CD38+HLA-DR+ CD4+ and CD8+ T-cells (supplemental Table 2), illustrating the increased activation of the T-cell compartment.
Both variants c.291A>G (p.Ile97Met) and c.302C>T (p.Thr101Ile) have been shown to abrogate TIM-3 expression.11-13 With the genetic identification of the 2 variants in our patient, TIM-3 expression was assessed on monocytes, B-cells, NK-cells, and T-cells. TIM-3 expression was absent from the patient’s monocytes and NK-cells ex vivo (Figure 2A-B). TIM-3 upregulation on activated T-cells after 3 days of stimulation with αCD3, αCD3/αCD28, or IL-15, was also absent in our patient, whereas CD25 expression was comparable to that of HDs (Figure 2C-D,F and data not shown).
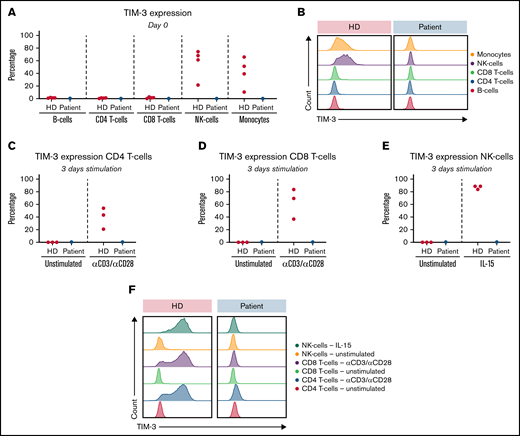
TIM-3 expression and upregulation in vitro. (A) Ex vivo TIM-3 expression on day 0: HD (red); patient (blue). (B) Representative histogram of TIM-3 expression on day 0. (C) TIM-3–expressing CD4 T-cells after 3 days of culture: HD (red); patient (blue). (D) TIM-3–expressing CD8 T-cells after 3 days of culture: HD (red); patient (blue). (E) TIM-3–expressing NK-cells after 3 days of culture: HD (red); patient (blue). (F) Representative histogram of TIM-3 expression after 3 days of culture.
TIM-3 expression and upregulation in vitro. (A) Ex vivo TIM-3 expression on day 0: HD (red); patient (blue). (B) Representative histogram of TIM-3 expression on day 0. (C) TIM-3–expressing CD4 T-cells after 3 days of culture: HD (red); patient (blue). (D) TIM-3–expressing CD8 T-cells after 3 days of culture: HD (red); patient (blue). (E) TIM-3–expressing NK-cells after 3 days of culture: HD (red); patient (blue). (F) Representative histogram of TIM-3 expression after 3 days of culture.
TIM-3 acts as a negative immune check point and is also a crucial regulator of innate immunity and inflammatory responses. It decreases interferon-γ–driven inflammation by suppressing TH1-cell responses.21,22 Therefore, it has been suggested that defective TIM-3 is responsible for macrophage activation and manifestations of hemophagocytosis.11 Apart from activated T-cells, TIM-3 is normally also expressed by activated NK-cells23 ; however, it was absent in our patient (Figure 2E-F). Because our patient was successfully treated, we measured NK-cell function again after 9 months and found that it had fully normalized (Figure 1D), whereas TIM-3 expression was still completely absent. These results confirm that cytotoxicity and degranulation of NK-cells are not dependent on TIM-3 expression.21 Notably, a follow-up demonstrated persistent lymphopenia (supplemental Table 2).
In summary, we describe a unique patient with features of dysregulated immune activation or proliferation /HLH without cytopenia, 2 HAVCR2 variants previously described in patients with HLH-SPTCL, but no T-cell lymphoma. The_c.291A>G (p.Ile97Met) and c.302C>T (p.Thr101Ile) compound heterozygosity results in absent TIM-3 expression, confirming the pathogenic nature of both variants, and together extends the clinical spectrum of this disease. As was proposed by Gayden et al,11 hemophagocytosis, because of the HAVCR2 variants, reflects an immune activation syndrome against which treatment with immunosuppressive agents could be sufficiently effective, whereas allo-HSCT is the current optimal treatment for classic primary HLH.
Acknowledgments: The authors thank the patient and healthy donors for participating in the study; Paul A. Baars and technicians of the Laboratory for Medical Immunology (Amsterdam UMC, location University of Amsterdam) for performance and analysis of immunological diagnostic tests; Geneviève de Saint Basile (Laboratory for Normal and Pathological Homeostasis of the Immune System, Hôpital Necker Enfants Malades, Paris, France) for help with evaluation of the patient.
S.A.M.T. was funded by the Center for Immunodeficiencies Amsterdam (grant 2015).
Contribution: S.A.M.T., M.A.G., L.K., E.M.M.v.L., S.J.B.M., and T.W.K. conceptualized the study; S.A.M.T. and M.H.J. performed the experiments; S.A.M.T., M.A.G., and T.W.K. wrote the original draft of the manuscript; C.E.R. and T.W.K. supervised the study; and all authors wrote, reviewed, and edited the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Samantha A. M. Tromp, Department of Experimental Immunology, Amsterdam UMC (location AMC), K0-140, Meibergdreef 9, 1105 AZ Amsterdam, The Netherlands; e-mail: s.a.tromp@amsterdamumc.nl.

