

Key Points
Obinutuzumab can be safely combined with venetoclax in patients with previously untreated follicular lymphoma in need of systemic therapy.
Despite the activity observed, our results do not support further development of the combination in this patient population.

Abstract
This phase 1 study evaluated safety, tolerability, and preliminary efficacy of obinutuzumab in combination with venetoclax in patients with previously untreated grade 1-3a follicular lymphoma in need of systemic therapy. Two DLs of venetoclax were evaluated with an expansion cohort at the recommended phase 2 dose. Twenty-five patients were enrolled. The recommended phase 2 dose was venetoclax 800 mg OD continuously for 6 cycles starting on day 2 of cycle 1, with obinutuzumab 1000 mg on days 1, 8, and 15 of cycle 1 and on day 1 of cycles 2 to 6, followed by obinutuzumab maintenance every 2 months for 2 years. Only 1 patient had a DLT consisting of grade 4 thrombocytopenia after the first obinutuzumab infusion. Neutropenia was the most common adverse event of grade ≥3 at least possibly attributed to study treatment. Twenty-four patients were evaluable for response after cycle 6 by computed tomography (CT) and 19 by positron emission tomography/CT (PET/CT): overall and complete response rates were 87.5% (95% CI, 67.6% to 97.3%) and 25% (95% CI, 9.8% to 46.7%) in the CT-evaluated patients and 84.2% (95% CI, 60.4% to 96.6%) and 68.4% (95% CI, 43.4% to 87.4%), respectively, in the PET/CT-evaluated patients. One-year progression-free survival was 77.8% (95% CI, 54.6% to 90.1%) and 79% (95% CI, 47.9% to 92.7%) for CT and PET/CT-evaluable patients, respectively, whereas progression-free survival at 30 months was 73.2% (95% CI, 49.8%, 87.0%) as assessed by CT and 79.0% (95% CI, 47.9%, 92.7%) by PET/CT. Despite the activity observed, our results do not support further development of the combination in this patient population. This trial was registered at www.clinicaltrials.gov as #NCT02877550.
Introduction
Follicular lymphoma (FL) is the most common indolent lymphoma. Most patients have an excellent long-term outcome, with median progression-free survival (PFS) up to 10 years and overall survival times approaching 2 decades.1 However, the disease is heterogeneous, and approximately 20% of patients progress within 2 years after first-line therapy and have a poor prognosis.2,3 One of the major achievements in the management of FL was the addition of rituximab to chemotherapy. Rituximab-based chemo-immunotherapy (bendamustine, or CHOP-like regimens) with or without rituximab maintenance is a standard first-line therapy for most patients with advanced disease.4-6
Over the last years, new monoclonal antibodies have been developed and tested in different lymphoma subtypes. Obinutuzumab, a type II, glycoengineered, humanized anti-CD20 monoclonal antibody combined with chemotherapy, resulted in improved PFS compared with rituximab-based chemo-immunotherapy in the GALLIUM phase 3 trial in previously untreated patients with FL.7 Based on results of this trial, binutuzumab has been approved in combination with chemotherapy in the first-line treatment of FL.
In addition to new monoclonal antibodies, several small molecules targeting components of intracellular pathways relevant in lymphomagenesis have also been clinically tested in different lymphoma subtypes, including FL.8,9
Many of these small molecules have subsequently entered clinical development in combination with anti-CD20 monoclonal antibodies, and there is currently high interest for the development of chemotherapy-free regimens, both for relapsed and treatment-naïve FL.10,11 One such small molecule, lenalidomide in combination with rituximab, was not inferior in comparison with rituximab and standard chemotherapy in a randomized phase 3 trial in untreated patients with FL.10
Venetoclax is a highly selective, potent, oral B-cell lymphoma 2 (BCL2) inhibitor with activity in different hematologic malignancies, including chronic lymphocytic leukemia, acute myeloid leukemia, multiple myeloma, and non-Hodgkin lymphomas.12-17 In the phase 1 trial in patients with relapsed or refractory non-Hodgkin lymphoma, venetoclax was well tolerated, and single-agent activity was observed in different lymphoma subtypes, including 38% responses among 29 patients with FL.17 With a median follow-up of 41.4 months (range, 7.7-0.5) for patients with FL, median PFS and duration of response were 10.8 months (95% CI, 5.6-16.0) and 26.6 months (95% CI, 3.1-45.6), respectively, being longer for patients achieving a complete response.18
Here we report results of a phase 1 trial of venetoclax in combination with obinutuzumab in patients with advanced FL in need of first-line systemic therapy.
Methods
Patient selection
Patients were eligible if they had histologically documented FL grade 1-3a, stage III-IV or stage II not suitable for radiotherapy and were in need of first-line systemic therapy defined as symptomatic disease, bulky disease (≥6 cm), clinically significant progression of any tumor lesion over at least 6 months, or B-symptoms (weight loss >10% in 6 months, drenching night sweats, fever >38°C not due to infection). Patients had to have measurable disease according to the Lugano classification.19
Other key eligibility criteria included Eastern Cooperative Oncology Group performance status 0 to 2; adequate hematologic, hepatic, and renal function (absolute neutrophil count ≥1.5 × 109/L; platelets ≥100 × 109/L or ≥75 × 109/L if BM involvement; aspartate aminotransferase [AST] and alanine aminotransferase and total bilirubin ≤1.5 times upper limit of normal; creatinine clearance >50 mL/min); and no prior systemic therapy. Patients with indolent lymphoma other than FL and those with FL grade 3b, known primary central nervous system lymphoma, or leptomeningeal involvement were excluded.
The institutional review board/ethics committees of participating centers approved the study. All patients provided written informed consent before enrollment. The study followed the ethical principles of the Declaration of Helsinki, the International Conference on Harmonization Guideline for Good Clinical Practice, and local regulations.
Study design, treatment administered, and patient evaluation
This was a multicenter, open-label, phase 1 study. The primary objective was to define the maximum tolerated dose and recommended phase 2 dose (RP2D) of venetoclax in combination with obinutuzumab in patients with previously untreated FL in need of systemic therapy. Secondary objectives included determination of safety, tolerability, and preliminary antitumor activity of the combination. Additional translational research included evaluation of minimal residual disease (MRD), BCL2 expression in lymphoma tissue, and liquid biopsy analysis for pretreatment genotype and treatment-associated clonal evolution.
Two dose levels (DLs) of venetoclax were evaluated with an expansion cohort at the RP2D. DL1 consisted of venetoclax 600 mg once daily (OD) and DL2 consisted of venetoclax 800 mg OD, administered orally, starting on day 2 of cycle 1, continuously for six 28-day cycles. The target dose of 800 mg was determined based on safety data from other combination studies that reached up to this dose as the RP2D.20,21 No ramp-up of venetoclax dose was foreseen in any of the DLs evaluated. In both DLs, patients received intravenous obinutuzumab 1000 mg on days 1, 8, and 15 of cycle 1 and on day 1 of cycles 2 to 6. Following cycle 6, patients with partial remission (PR) or complete remission (CR) continued maintenance treatment with single-agent obinutuzumab 1000 mg every 2 months for 2 years, similar to other trials in first-line treatment of FL.7
Dose escalation followed the standard 3 + 3 design.22 The RP2D was defined as the DL in which ≤1 of 6 patients developed dose-limiting toxicity (DLT).
Toxicity was graded using the National Cancer Institute Common Terminology Criteria for Adverse Events version 4.03. DLTs were defined as adverse events occurring during the first 28-day cycle of treatment and fulfilling 1 of the following criteria: absolute neutrophil count <0.5 × 109/L for 8 days or longer; febrile neutropenia; platelets <25 × 109/L or thrombocytopenic bleeding (platelets <50 × 109/L and associated with clinically significant bleeding); any grade 3 or higher nonhematologic toxicity at least possibly related to trial treatment except alopecia, nausea, and diarrhea adequately treated; any grade 3 or higher laboratory abnormalities lasting for >7 days; any grade 3 laboratory changes consistent with tumor lysis syndrome (TLS) if associated with manifestations of clinical TLS (increased creatinine, hyperuricemia, hyperkalemia, hypophosphatemia, hypocalcemia); trial therapy–related death; ≥8 missed therapy days of venetoclax due to trial drug–related toxicity; ≥2 missed doses of obinutuzumab due to trial drug–related toxicity; delay of >3 weeks of cycle 2 due to trial drug–related toxicity.
Response was assessed with contrast-enhanced CT or PET/CT or total body magnetic resonance imaging (MRI) using the Lugano criteria after 3 and 6 cycles during combination therapy and after 6, 12, and 24 months of obinutuzumab maintenance. PET/CT images were made available in a digitized form for independent response review. A BM biopsy was performed only if clinically indicated and in patients with BM involvement at baseline in case of CT or PET/CT complete response.
Supportive medications and dose modifications
Appropriate hydration and administration of an agent to reduce uric acid were required before start of treatment. Patients considered to be at high risk of developing TLS (any mass ≥10 cm, circulating lymphoma cells) were hospitalized for more intensive prophylaxis and monitoring during the initial dose of venetoclax. After adverse events observed in 1 patient after the first obinutuzumab infusion, the protocol was amended to permit a prephase with steroids (50-100 mg prednisone per day) for up to 5 days before start of treatment in patients with high tumor burden (any mass ≥10 cm, circulating lymphoma cells, high BM infiltration) based on investigator’s decision. Antibacterial prophylaxis was recommended and left to the investigator’s discretion. Preventive use of growth factor support was not allowed during cycle 1 and could be used based on local policy thereafter.
Study drugs were withheld for neutropenia grade ≥4, thrombocytopenia grade ≥3, and nonhematologic adverse events grade ≥3. No dose reductions were permitted for obinutuzumab, whereas venetoclax dose could be reduced to 400 mg because lower doses were considered ineffective in FL based on the single-agent phase 1 trial.17
Duration of study treatment
Patients who had less-than-minimal response (defined as >25% decrease from baseline in their sum of product of diameter of up to 6 dominant, measurable nodes and extranodal sites) after 3 cycles and less than a PR after 6 cycles were discontinued (unless they were staged by CT/PET and had metabolic response). During maintenance with obinutuzumab, patients with response or stable disease were allowed to continue up to 2 years (12 administrations) or until disease progression. Symptomatic deterioration, unacceptable adverse events, patient decision to withdraw consent from the study, treatment delay for >3 weeks, or pregnancy were other reasons for treatment interruption.
Statistical analysis.
All efficacy analyses were based on the full analysis set defined as all registered patients who received at least 1 dose of trial treatment (both drugs), excluding patients with major eligibility violations described in the statistical analysis plan. All safety analyses were based on the safety analysis set defined as all patients who received any dose of trial treatment. In general, the summary statistics presented for quantitative variables were the median, minimum, and maximum values. For each endpoint related to objective tumor responses, the proportion of patients and associated 95% CI (Clopper Pearson) were presented. The summary statistics presented for categorical data were the count and percentage of patients in each category. Median PFS, median DoR, and PFS at 12 and 30 months were calculated using the Kaplan-Meier method with its corresponding 95% CI. No statistical testing was performed. These endpoints were presented separately for each of the imaging methods. All analyses were performed in SAS 9.4 (SAS Institute Inc., Cary, NC) and R version 4.0.3 (Foundation for Statistical Computing, Vienna, Austria).
Translational research
Central BCL2 expression.
Immunohistochemistry with clone E17 and SP66 was performed in a central lymphoma pathology reference center according to standard procedures in formalin-fixed paraffin embedded tumor tissue.23
MRD evaluation.
Bone marrow (BM) and peripheral blood (PB) samples were analyzed in a central laboratory by polymerase chain reaction (PCR) at baseline for the presence of the BCL2/IgH gene fusion to be used as molecular markers during follow-up. DNA was isolated using the DNeasy Blood & Tissue Kits (Qiagen, Hilden, Germany). PCR for t(14;18)(q32;q21) was performed with the IdentiClone BCL2/JH Translocation Assay (InVivoScribe Technologies, San Diego, CA) based on the BIOMED-2 collaborative study and with a sensitivity between 3.3 × 10−2 and 10−3.24 The assay encompasses the Mbr, the 3′MBr, and the mcr regions. Only samples positive at baseline were analyzed at the end of the first 6 cycles of treatment and in case of relapse. Samples were scored according to the kit manufacturers’ guidelines. Briefly, at baseline, a sample was scored positive when a PCR product could be demonstrated within the size range expected from the set of primers in use. A sample was scored negative when it did not show a PCR product within the size range expected from the set of primers in use, in the presence of a positive control, and if the same DNA sample had given the expected products using a set of primers to evaluate its quality. For follow-up samples, a sample was scored positive when it showed a PCR product of the same size as at baseline; a sample was scored negative as defined at baseline.
Liquid biopsies.
Blood samples for liquid biopsies were collected from a limited number (n = 4) of patients participating in this trial and from 2 patients who previously gave consent for an ancillary study on liquid biopsies active at the Oncology Institute of Southern Switzerland (upon agreement of the sponsor and the lead ethics committee). Blood samples consisted of 20 mL of PB in EDTA tubes and 20 mL of PB in cell-free DNA BCT tubes at baseline, after 3 and 6 cycles of combination therapy, in additional disease evaluations when a PET/CT was performed, and in case of progression.
LyV4.0 CAncer Personalized Profiling (CAPP) by deep sequencing assay was used for the study. Libraries derived from paired germline genomic DNA and cell-free DNA from the same patient were sequenced simultaneously to avoid batch-related biases, using the NextSeq500 (Illumina, San Diego, CA) instrument by pair-end sequencing (2 × 150 cycle protocol). A total of 26 multiplexed libraries were sequenced, in 2 separated ultra-deep experiments.
Variants for the resulting candidate mutations and the background error rate were visualized using Integrative Genomics Viewer.
Central PET-CT review.
For the present study, all the PET/CT scans were centrally evaluated following a standard protocol with dedicated imaging software (MM Oncology, Syngo.via, Siemens). PET scans obtained at baseline and after 6 months of treatment were first qualitatively analyzed, and the residual metabolic findings were graded applying the Deauville 5-point scale.25 The achievement of a complete metabolic response was defined according to the criteria of the Lugano classification by a completely PET-negative scan or a scan having minimal residual uptake less than the liver activity (Deauville score 1-3).19
For patients who had liquid biopsies, lymphoma lesions were automatically segmented by fixing a standardized uptake value (SUV) of 4 as threshold and the metabolic tumor volume (MTV) was estimated.26,27
Results
Patient characteristics
Between March 2017 and May 2019, 25 patients with FL were included into the trial at 6 Swiss and 2 German participating sites. One patient was not treated with venetoclax. This patient was included in the safety analysis set used for safety evaluations but excluded from the full analysis set used for efficacy evaluations, as predefined by the statistical analysis plan. Baseline characteristics of the patients are reported in Table 1. Most patients had advanced disease (92% had stage III or IV disease) and 12 (48%) had FLIPI high risk, while the main reason for starting systemic therapy was symptomatic disease followed by clinically significant progression of disease in 6 months.
Baseline patient characteristics
| Characteristic | Total N= 25 n (%) | |
|---|---|---|
| Median age, y (range): 55 (30-78) | ||
| Sex | Male | 13 (52) |
| ECOG | 0 | 21 (84) |
| 1 | 3 (12) | |
| 2 | 1 (4) | |
| Ann Arbor Stage | II | 2 (8) |
| III/IV | 23 (92) | |
| FL grade | 1 or 2 | 19 (76) |
| 3a | 6 (24) | |
| FLIPI | Low risk (0-1) | 5 (20) |
| Intermediate risk (2) | 8 (32) | |
| High risk (3-5) | 12 (48) | |
| Reason for treatment start (>1 possible) | B-symptoms | 10 (40) |
| Bulky disease (≥6 cm) | 11 (44) | |
| Progression in the last 6 mo | 13 (52) | |
| Symptomatic disease | 20 (80) | |
| Characteristic | Total N= 25 n (%) | |
|---|---|---|
| Median age, y (range): 55 (30-78) | ||
| Sex | Male | 13 (52) |
| ECOG | 0 | 21 (84) |
| 1 | 3 (12) | |
| 2 | 1 (4) | |
| Ann Arbor Stage | II | 2 (8) |
| III/IV | 23 (92) | |
| FL grade | 1 or 2 | 19 (76) |
| 3a | 6 (24) | |
| FLIPI | Low risk (0-1) | 5 (20) |
| Intermediate risk (2) | 8 (32) | |
| High risk (3-5) | 12 (48) | |
| Reason for treatment start (>1 possible) | B-symptoms | 10 (40) |
| Bulky disease (≥6 cm) | 11 (44) | |
| Progression in the last 6 mo | 13 (52) | |
| Symptomatic disease | 20 (80) | |
At the time of data cutoff (22 March 2022), 15 patients had completed treatment as per protocol, 7 discontinued due to progressive disease (2 during combination therapy and 5 during obinutuzumab maintenance), 2 were discontinued for principal investigator decision (1 due to adverse events observed after the first obinutuzumab infusion and 1 during maintenance following COVID-19 infection), and 1 patient withdrew consent during maintenance.
DLTs and recommended phase 2 dose
Only 1 patient treated at DL2 had a DLT consisting of grade 4 thrombocytopenia after the first obinutuzumab infusion. Therefore, the RP2D of the combination is venetoclax 800 mg OD continuously for 6 cycles starting on day 2 of cycle 1 with obinutuzumab 1000 mg on days 1, 8, and 15 of cycle 1 and on day 1 of cycles 2 to 6, followed by obinutuzumab maintenance for up to 2 years.
Safety and compliance
No deaths were reported. Most frequent adverse events of at least possible attribution to study treatment are described in Table 2. No cases of TLS were observed. Fatigue and infusion-related reactions were the most frequent and were all grade 1 or 2. Among other nonhematologic adverse events, nausea and diarrhea were reported each in 9 patients (36%) and were grade 1 or 2 except for 1 patient presenting with diarrhea grade 3 (another patient had diarrhea grade 2 and 7 patients had grade 1). Neutropenia (in 36% of patients; 20% grade 3 and 8% grade 4) and thrombocytopenia (28%; 4% grade 3 and 4% grade 4) were the most frequent hematologic adverse events. Only 1 patient was discontinued based on the decision of the principal investigator following the development of adverse events after the first obinutuzumab infusion consisting of thrombocytopenia grade 4, pneumonitis grade 3, and AST increase grade 3. These events were all considered related to a reaction after the obinutuzumab infusion (this patient never received venetoclax) and were successfully managed with intravenous steroids.
Adverse events of at least possible attribution to study treatment present in at least 8% of patients (including laboratory abnormalities considered clinically significant by the treating investigator)
| Event, n (%) | All grades | ≥ Grade 3 |
|---|---|---|
| Fatigue | 12 (48) | 0 |
| Infusion related reaction | 11 (44) | 0 |
| Nausea | 9 (36) | 0 |
| Diarrhea | 9 (36) | 1 (4) |
| Neutropenia | 9 (36) | 7 (28) |
| Thrombocytopenia | 7 (28) | 2 (8) |
| Fever | 6 (24) | 0 |
| Anemia | 5 (20) | 2 (8) |
| Constipation | 4 (16) | 0 |
| Leukopenia | 3 (12) | 0 |
| Dysgeusia | 3 (12) | 0 |
| Chills | 3 (12) | 0 |
| Febrile neutropenia | 2 (8) | 2 (8) |
| Vomiting | 2 (8) | 0 |
| Upper respiratory/bronchial infection | 2 (8) | 2 (8) |
| AST increase | 2 (8) | 1 (4) |
| Creatinine increase | 2 (8) | 0 |
| Lymphopenia | 2 (8) | 1 (4) |
| Anorexia | 2 (8) | 0 |
| Dizziness | 2 (8) | 0 |
| Rash | 2 (8) | 0 |
| Event, n (%) | All grades | ≥ Grade 3 |
|---|---|---|
| Fatigue | 12 (48) | 0 |
| Infusion related reaction | 11 (44) | 0 |
| Nausea | 9 (36) | 0 |
| Diarrhea | 9 (36) | 1 (4) |
| Neutropenia | 9 (36) | 7 (28) |
| Thrombocytopenia | 7 (28) | 2 (8) |
| Fever | 6 (24) | 0 |
| Anemia | 5 (20) | 2 (8) |
| Constipation | 4 (16) | 0 |
| Leukopenia | 3 (12) | 0 |
| Dysgeusia | 3 (12) | 0 |
| Chills | 3 (12) | 0 |
| Febrile neutropenia | 2 (8) | 2 (8) |
| Vomiting | 2 (8) | 0 |
| Upper respiratory/bronchial infection | 2 (8) | 2 (8) |
| AST increase | 2 (8) | 1 (4) |
| Creatinine increase | 2 (8) | 0 |
| Lymphopenia | 2 (8) | 1 (4) |
| Anorexia | 2 (8) | 0 |
| Dizziness | 2 (8) | 0 |
| Rash | 2 (8) | 0 |
A total of 18 serious adverse events occurred in 11 patients, 9 of them considered at least possibly related to study treatment as assessed by investigator and sponsor. They consisted of febrile neutropenia G3 (n = 2), and 1 event each for fever G1, bronchial infection G3, upper respiratory infection G3, AST G3, thrombocytopenia grade 4, pneumonitis grade 3, and COVID-19 infection.
The median relative dose intensity (proportion of administered doses relative to planned doses) was 100% for venetoclax and 96.6% for obinutuzumab.
Antitumor activity
Twenty-four patients were evaluable for response at 6 months by CT: CR rate was 25% (95% CI, 9.8% to 46.7%) and overall response rate (ORR) was 87.5% (95% CI, 67.6% to 97.3%). Nineteen patients were evaluated by PET/CT: CR rate was 68.4% (95% CI, 43.4% to 87.4%) and ORR was 84.2% (95% CI, 60.4% to 96.6%) (Table 3). By central PET review, 14 patients had CR (73.7%) and 2 had PR, for an ORR of 84.2%.
CT and PET-CT response assessment at 6 months (end of combination therapy)
| Method of assessment | N evaluable patients | CR, % | 95% CI (Clopper Pearson) | ORR, % | 95% CI (Clopper Pearson) |
|---|---|---|---|---|---|
| CT | 24 | 25.0 | (9.8; 46.7%) | 87.5 | (67.6; 97.3%) |
| PET/CT | 19 | 68.4 | (43.4; 87.4%) | 84.2 | (60.4; 96.6%) |
| Method of assessment | N evaluable patients | CR, % | 95% CI (Clopper Pearson) | ORR, % | 95% CI (Clopper Pearson) |
|---|---|---|---|---|---|
| CT | 24 | 25.0 | (9.8; 46.7%) | 87.5 | (67.6; 97.3%) |
| PET/CT | 19 | 68.4 | (43.4; 87.4%) | 84.2 | (60.4; 96.6%) |
At 30 months, CR rate assessed by CT was still 25.0% (95% CI, 9.8% to 46.7%) and ORR was 62.5% (40.6% to 81.2%). According to PET/CT, CR and ORR at 30 months were 52.6% (28.9% and 75.6%, respectively). ORR based on best response during whole therapy (combination and maintenance) was 87.5% (95% CI, 67.6% to 97.3%) as assessed by CT/MRI and 89.5% (95% CI, 66.9% to 98.7%) as assessed by PET/CT.
Median time to follow-up was 28.1 months (95% CI, 18.9-28.7).
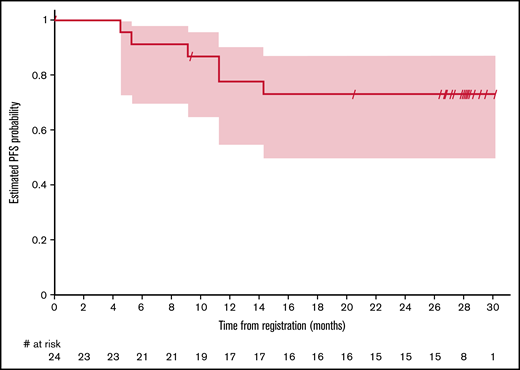
One-year PFS was 77.8% (95% CI, 54.6% to 90.1%) and 79.0% (95% CI, 47.9% to 92.7%) for CT and PET/CT evaluable patients, respectively (Figure 1). PFS at 30 months was 73.2% (95% CI, 49.8% to 87.0%) as assessed by CT and 79.0% (95% CI, 47.9% to 92.7%) as assessed by PET/CT. Median PFS and duration of response were not reached.
BCL2, MRD, liquid biopsies, and PET-metrics
BCL2 expression.
Nineteen patients had tissue available for central immunohistochemical analysis of BCL2 expression by immunohistochemistry with antibodies directed against the epitopes E17 and SP66. E17 was positive in all patients (weak positivity in 1 patient who tested positive for SP66), whereas SP66 was positive in 17 patients and negative in 2 patients. This discrepancy can be explained by missense mutations of the BCL2 gene, suggesting that the BCL2 protein “pseudonegativity” in these 2 cases is the result of structural protein modifications that interfere with the binding of the BCL2 antibody.23 Both patients had complete metabolic response at 6 months. Thus, it can be concluded that all cases were typical FL with constitutive BCL2 expression.
MRD evaluation.
The presence of the t(14;18) by PCR was assessed at baseline in PB from 24 patients and in the BM from 19 patients. Nine patients had a positive signal at baseline both at BM and at PB. In addition, among 5 patients who had only PB assessed, 3 had a positive signal. Of the 9 patients with a baseline signal both at PB and BM, 6 had MRD evaluated at 6 months (end of combination therapy) both in BM and PB, and all were negative. All 6 patients had CR by PET/CT (5 PR and 1 CR by CT only). In 3 patients, only PB was assessed after the end of combination therapy: MRD was negative in 2 patients (both had PR by CT and CR by PET) and positive in 1 patient (PD by CT). Among 3 patients who had only PB assessed at baseline, all were negative after 6 cycles of trial treatment (PR by CT and PET in 1, CR by CT and PET in 1, PR by CT and CR by PET in 1 patient).
Liquid biopsies.
In total, 20 cell-free DNA samples from 6 patients were analyzed. Our LyV4.0 ctDNA CAPP-seq assay targeted ∼344 kb of genomic space and was designed to cover a variety of coding genomic regions known to be recurrently mutated in mature BCLs plus coding/noncoding region targeted by aberrant somatic hypermutation. The limit of quantification of the LyV4.0 ctDNA CAPP-seq assay was 0.09%, and the analytical sensitivity was 0.1%. The molecular profiles documented in pretreatment ctDNA were consistent with previously reported genetic signatures in untreated FL. Quantifiable levels of ctDNA (Figure 2) disappeared under treatment in 3 of 6 patients, whereas they persisted in the remaining 3 cases. Only 1 patient (among those with persistent quantifiable levels of DNA) had a clinical and radiologic early disease progression after cycle 5 and was removed from the trial due to progression. All other patients were radiologically responding and on maintenance therapy at the time of data cutoff. In 5 patients with liquid biopsies, a PET scan was also available, and changes in MTV correlated with changes in levels of ctDNA (Figure 3).
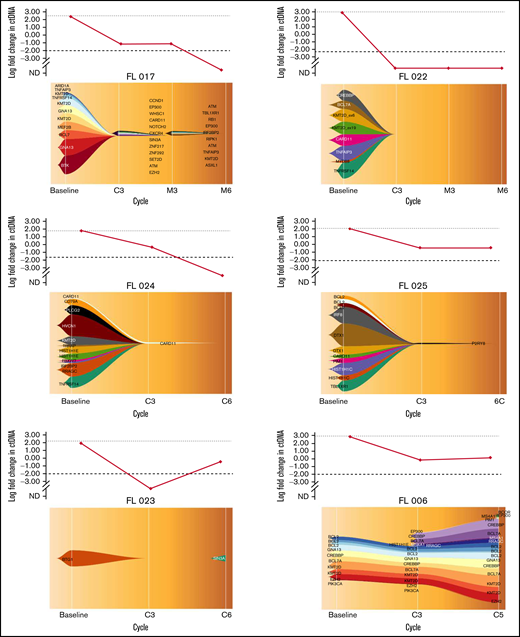
Clonal evolution with the fishplot package in 6 patients. The vertical axis of the upper graph represents the log fold change in ctDNA load. The horizontal axis of both upper graph and fishplot shows the treatment timepoints. The fishplot shows the clonal evolution across the timepoints, with the list of mutated genes detected, represented as descent in the form of parental relationships. Patients FL 017, FL 022, and FL 024 had quantifiable levels of ctDNA that disappeared under treatment. Patients FL 025, DFL023, and FL 006 had quantifiable levels of ctDNA that persisted under treatment. Only patient FL 006 had a clinical and radiological progression of disease detected after cycle 5. Imaging in this patient was performed before end of cycle 6 due to an increase of LDH levels. C, cycle; EoM, end of maintenance; M, maintenance.
Clonal evolution with the fishplot package in 6 patients. The vertical axis of the upper graph represents the log fold change in ctDNA load. The horizontal axis of both upper graph and fishplot shows the treatment timepoints. The fishplot shows the clonal evolution across the timepoints, with the list of mutated genes detected, represented as descent in the form of parental relationships. Patients FL 017, FL 022, and FL 024 had quantifiable levels of ctDNA that disappeared under treatment. Patients FL 025, DFL023, and FL 006 had quantifiable levels of ctDNA that persisted under treatment. Only patient FL 006 had a clinical and radiological progression of disease detected after cycle 5. Imaging in this patient was performed before end of cycle 6 due to an increase of LDH levels. C, cycle; EoM, end of maintenance; M, maintenance.
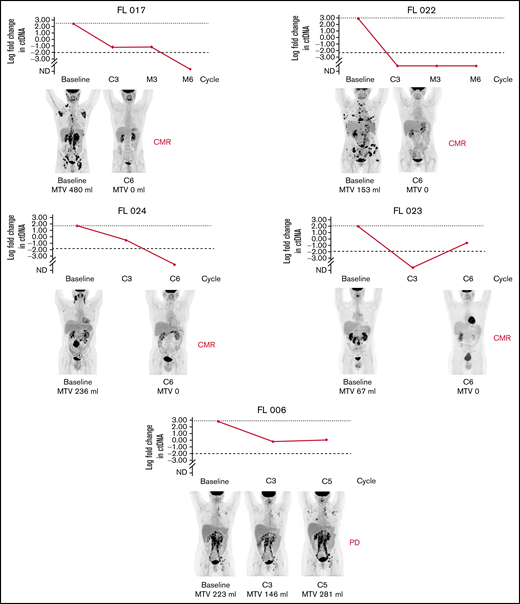
Correlation of clonal evolution and MTV in 5 patients with both data available. Only patient 006 had evidence of progressive disease by PET. C, cycle; CMR, complete metabolic remission; EoM, end of maintenance; M, maintenance; PD, progressive disease.
Correlation of clonal evolution and MTV in 5 patients with both data available. Only patient 006 had evidence of progressive disease by PET. C, cycle; CMR, complete metabolic remission; EoM, end of maintenance; M, maintenance; PD, progressive disease.
Discussion
The BCL2 inhibitor venetoclax and the type II anti-CD20 monoclonal antibody obinutuzumab are 2 anti-lymphoma drugs that have entered in the standard of care for selected hematologic malignancies. In previously untreated patients with FL, obinutuzumab in combination with chemotherapy has shown superior PFS and a marked reduction in the rate of early disease progression in comparison with rituximab plus chemotherapy.7,28 On the other hand, the overexpression of BCL2 in virtually all cases of FL provides a strong rationale for the use of venetoclax in this disease.29
Aiming at developing a chemotherapy-free regimen, we performed a phase 1 trial combining obinutuzumab with venetoclax for 6 months, followed by obinutuzumab maintenance for up to 2 years, in patients with previously untreated FL in need of systemic therapy. The 2 drugs could be safely combined, and the recommended phase 2 dose was established at venetoclax 800 mg OD continuously starting on day 2 of cycle 1 with obinutuzumab 1000 mg on days 1, 8, and 15 of cycle 1 and on day 1 of cycles 2 to 6. The observed toxicities were predictable and manageable, and patients were able to receive treatment as per protocol without any significant dose delays or interruptions. Only 1 patient had to discontinue treatment based on principal investigator decision following an exceptional response and adverse events observed after the first dose of obinutuzumab. No cases of TLS were observed, and venetoclax was administered starting on day 2 without any need for ramp-up dose. Most patients responded to treatment; the rate of CR at 30 months by PET/CT was 52.6% and PFS at 30 months was 79.0% as assessed by PET/CT. Additional endpoints, including MRD evaluation, liquid biopsies for assessment of molecular responses and correlation with PET results in a small number of patients tested, and BCL2 expression, were performed, but their results did not permit identification of any new markers of response because the small number of included patients made them uninformative.
Our study was not designed to test the efficacy of venetoclax with obinutuzumab in previously untreated patients with FL, nor is it possible to draw any conclusion regarding any potential benefit that could derive by adding venetoclax to anti-CD20 therapy in this setting. Obinutuzumab single agent resulted in responses up to 66.2% (best overall response during induction and maintenance) in patients with relapsed FL.30 On the other hand, the activity of venetoclax in FL is not clear yet. Before our trial, the Contralto trial evaluated venetoclax in combination with either rituximab or rituximab and bendamustine vs bendamustine and rituximab in previously treated patients with FL.31 This study did not show a benefit by adding venetoclax to any of the treatment arms; however, authors concluded that there was evidence that some patients in the bendamustine-free arm could have benefited.
The observed 1-year and 30-month PFS were lower if compared with other anti-CD20–based combinations; thus, we were not able to provide any evidence of benefit by adding venetoclax to obinutuzumab in this setting. It remains unknown if a longer treatment duration with venetoclax could have resulted in better results. However, when we designed our trial, there was not enough information on the long-term safety or efficacy of venetoclax in patients with treatment-naive FL, and we decided to use a fixed duration of 6 months of venetoclax as part of induction therapy and continue with only obinutuzumab maintenance in responding patients. A phase 2 trial with a longer venetoclax administration or a treatment duration based on metabolic and/or molecular response would have been more informative in that sense, but currently there is no plan for such a trial. Another ongoing clinical trial (#NCT03980171) is evaluating the combination of venetoclax with lenalidomide and obinutuzumab, and results are awaited.
Chemotherapy-free regimens have been developed for patients with FL and they could represent a valid alternative to chemotherapy combinations in patients in need of systemic therapy. One such combination of rituximab with lenalidomide was not inferior to rituximab and chemotherapy; therefore, it does represent a valid treatment option for newly diagnosed patients with advanced disease.10 Other combinations of molecularly targeted agents with anti-CD20 monoclonal antibodies may warrant evaluation in the first-line treatment of advanced FL.
In conclusion, we report results of a phase 1 trial of venetoclax combined with obinutuzumab in untreated FL, defining the recommended phase 2 dose. Although the 2 drugs could be safely combined and no new safety signals were identified, our results do not provide any clear benefit by adding venetoclax to anti-CD20 monoclonal antibody in patients with previously untreated FL. Addition of translational research questions to clinical endpoints, including the identification of mutations resulting in BCL2 inhibitor resistance,32 should be integrated in the design of future trials exploring the role of BCL2 inhibition in FL, which currently remains unclear.
Acknowledgments
This study was financially supported by F. Hoffmann-La Roche Ltd., the Swiss State Secretariat for Education, Research and Innovation (SERI), and the Swiss Cancer Research Foundation (SCS).
Authorship
Contribution: A.S., C.S., S.H., E.Z., and W.H. conceived and designed the study; A.S., D.R., S.D., and E.Z. developed the methodology; A.S., U.M., F.H., C.P., N.M., F.K., U.N., C.S., K.H., D.K., D.H., A.A.M., G.F., D.R., L.C., G.S., and F.B. acquired the data; A.S., U.M., S.S., F.K., U.N., S.H., D.R., S.D., L.C., E.Z., and W.H. analyzed and interpreted the data; A.S., U.M., S.S., C.P., F.K., U.N., C.S., D.K., D.H., A.A.M., S.H., G.F., D.R., S.D., L.C., F.B., C.B., E.Z., and W.H. wrote, reviewed, and/or revised the manuscript; F.H., C.S., K.E., and M.U. provided administrative, technical, or material support; and A.S., U.N., F.H., K.E., and W.H. provided study supervision.
Conflict-of-interest disclosure: A.S. served as consultant to Bayer and Eli Lilly and advisor to Roche and Janssen, received institutional research funding from Roche, AbbVie, Pfizer, Bayer, Merck, Novartis, ADC Therapeutics, and MEI Therapeutics, and received travel grants from AbbVie and PharmaMar. U.M. provided consultancy services to Roche and AbbVie and received travel grants from AbbVie and Roche. F.H. served on the advisory board for Roche and AbbVie. U.N. has served on the scientific advisory board since 2018 for Janssen-Cilag, Celgene (BMS), Takeda, AstraZeneca, Roche, Novartis, Gilead, and Pierre Fabre, received speakers honoraria from Celgene (BMS) and Novartis, and made contributions to congress fees for Novartis, Celgene (BMS), Amgen, Roche, and Takeda. C.S. received honoraria from Novartis, Kite Gilead, served as consultant for Novartis, Kite Gilead, Takeda, and BMS, received research funding from Bayer Healthcare and Kite Gilead, and received travel grants from Novartis, Janssen, Kite Gilead, BMS, and Takeda. D.S. is a shareholder of Roche. A.A.M. has served on the advisory board of Roche, Janssen, and Takeda. D.R. received honoraria from AbbVie, AstraZeneca, and Janssen and received research grants from AbbVie, AstraZeneca, and Janssen. S.D. served as advisor for Roche, Novartis, and Takeda. W.H. provided consultancy services to Roche and Janssen, served as advisor for Roche, and received institutional research funding from Roche and Janssen. F.B. received institutional research funds from Acerta, ADC Therapeutics, Bayer AG, Cellestia, CTI Life Sciences, EMD Serono, Helsinn, ImmunoGen, Menarini Ricerche, NEOMED Therapeutics 1, Nordic Nanovector ASA, Oncology Therapeutic Development, and PIQUR Therapeutics AG, received consultancy fees from Helsin and Menarini, provided expert statements to HTG, and received travel grants from Amgen, AstraZeneca, Jazz Pharmaceuticals, and PIQUR Therapeutics AG. E.Z. provided consultancy services to Beigene, Celgene, Incyte, Janssen, Merck, Roche, Celltion Healthcare, and Kite, received institutional research funding from AstraZeneca, Celgene, Incyte, Janssen, Merck, and Roche, and received travel grants from Roche. W.H. provided consultancy services to Roche and Janssen, provided advisory services to Roche, and received institutional research funding from Roche and Janssen. The remaining authors declare no competing financial interests.
Correspondence: Anastasios Stathis, Oncology Institute of Southern Switzerland, Ospedale Regionale di Bellinzona e Valli, Via A. Gallino 12, CH-6500 Bellinzona, Switzerland; e-mail: anastasios.stathis@eoc.ch.

