

Key Points
Hypersialylation in MM facilitates immune evasion of NK cells but can be overcome by targeted desialylation or genetic deletion of Siglec-7.
Desialylation unmasks CD38 expression on MM cells, enhancing NK cell–mediated ADCC induced by CD38 targeting of monoclonal antibodies.

Abstract
Abnormal glycosylation is a hallmark of cancer, and the hypersialylated tumor cell surface facilitates abnormal cell trafficking and drug resistance in several malignancies, including multiple myeloma (MM). Furthermore, hypersialylation has also been implicated in facilitating evasion of natural killer (NK) cell–mediated immunosurveillance but not in MM to date. In this study, we explore the role of hypersialylation in promoting escape from NK cells. We document strong expression of sialic acid-derived ligands for Siglec-7 (Siglec-7L) on primary MM cells and MM cell lines, highlighting the possibility of Siglec-7/Siglec-7L interactions in the tumor microenvironment. Interactomics experiments in MM cell lysates revealed PSGL-1 as the predominant Siglec-7L in MM. We show that desialylation, using both a sialidase and sialyltransferase inhibitor (SIA), strongly enhances NK cell–mediated cytotoxicity against MM cells. Furthermore, MM cell desialylation results in increased detection of CD38, a well-validated target in MM. Desialylation enhanced NK cell cytotoxicity against CD38+ MM cells after treatment with the anti-CD38 monoclonal antibody daratumumab. Additionally, we show that MM cells with low CD38 expression can be treated with all trans-retinoic acid (ATRA), SIA and daratumumab to elicit a potent NK cell cytotoxic response. Finally, we demonstrate that Siglec-7KO potentiates NK cell cytotoxicity against Siglec-7L+ MM cells. Taken together, our work shows that desialylation of MM cells is a promising novel approach to enhance NK cell efficacy against MM, which can be combined with frontline therapies to elicit a potent anti-MM response.
Introduction
Aberrant glycosylation is a hallmark of cancer. In contrast to nontransformed cells, the outer surface of tumor cells is frequently coated with a dense layer of sialic acids (hypersialylation), attached to glycoproteins and lipids by a family of sialyltransferases.1,2 Sialyltransferase expression is frequently dysregulated in cancer, and several sialyltransferases, including ST3GAL6, are implicated in disease progression and survival in multiple myeloma (MM).3,4 In addition to facilitating abnormal cell trafficking and drug resistance, hypersialylation may contribute to immune evasion, including suppression of natural killer (NK) cell–mediated immunosurveillance.5,6
NK cells are cytotoxic lymphocytes with an innate ability to recognize and eliminate malignant cells.7 They do this by employing a wide range of inhibitory and activating receptors.8,9 If a strong net activating signal is generated after engagement of these receptors upon interaction with a target cell, the NK cell is activated and the target cell is destroyed. In contrast, if an overall inhibitory signal is generated, the target cell is released without action. While self-recognition mediated via binding of human leukocyte antigen (HLA) class I molecules to the inhibitory receptors CD94/NK 2 group member A (NKG2A), killer cell immunoglobulin-like receptors (KIRs) and leukocyte Ig-like receptor (LIR-1) is well recognized, the potential for sialylated glycans to function as self-associated molecular patterns has only recently begun to be understood.10-12
Certain sialylated glycans on tumor cells can bind to sialic acid-binding immunoglobulin-like lectins (Siglecs), a family of receptors expressed predominantly by immune cells.13 Analogous to checkpoint receptors such as PD-1, upon engagement with their cognate ligands, inhibitory Siglecs recruit and activate intracellular phosphatases SHP-1 and SHP-2 through their intracellular immunoreceptor tyrosine-based inhibitory motif.14 Upon activation, these phosphatases regulate activation pathways inhibiting cytotoxicity and/or cytokine production.14,15 Only 2 members of the Siglec family are expressed by NK cells: inhibitory receptors Siglec-7 and Siglec-9. Whereas Siglec-7 is expressed on almost all NK cells, Siglec-9 is restricted to a subset of CD56dim NK cells.16,17 The expression of these receptors suggests that NK cells may be susceptible to inhibition in the presence of cancer cells expressing the cognate ligands. Indeed, ligands for both Siglec-7 and Siglec-9 are expressed on a wide range of cancer cell types.18-20 While these ligands have yet to be fully elucidated in MM, a recent study identified CD43 as the predominant ligand for Siglec-7 in chronic myelogenous leukemia.21 Finally, tumor desialylation using a sialidase has been shown to enhance NK cell–mediated cytotoxicity against several cancer types, and blockade of Siglec-7 and Siglec-9 has been demonstrated to enhance NK cell–mediated cytotoxicity.18,22
Based on this background, we hypothesized that hypersialylation may dampen the ability of NK cells to target MM cells. Therefore, we set out to address whether hypersialylation mediates NK cell immune evasion in MM, the potential role of Siglecs in facilitating this, and the identity of their ligands. We also aimed to determine whether strategies to remove sialic acids or target Siglecs could enhance NK cell–mediated cytotoxicity. Herein, we report that hypersialylation protects MM cells from NK cell immunosurveillance mediated, at least in part, by Siglec-7. Furthermore, we uncover that desialylation of MM cells also unmasks CD38, suggesting a novel dual role for tumor cell desialylation when combined with CD38-targeted therapies for MM. Targeting the sialic acid-Siglec axis may represent a novel glyco-immune checkpoint inhibitory strategy capable of enhancing NK cell–based immunotherapies in MM.
Materials and methods
Cell lines and MM patient-derived primary samples
Cell lines MM1S, JJN3, H929, K562, and NK-92 were from American Type Culture Collection (ATCC). KHYG-1 was kindly provided by Dr. Armand Keating (University of Toronto). All cell lines except NK-92 were cultured in RPMI-1640 supplemented with 10% heat-inactivated FBS (Sigma Aldrich), 100 IU/mL of penicillin, and 100 µg/mL of streptomycin. NK-92 cells were cultured in X-VIVO 10 media (Lonza) supplemented with 20% human AB serum (Sigma) and 500 IU/mL of interleukin (IL) 2. KHYG-1 cells were supplemented with 100 IU/mL of IL-2 (Peprotech). Fresh bone marrow (BM) was obtained from patients with MM after informed consent. Mononuclear cells (MNCs) were obtained from BM aspirates (BMAs) after density gradient centrifugation with Ficoll-Paque. CD138+ MM cells were isolated by magnetic bead-based positive selection (StemCell Technologies). MNC, CD138-, and CD138+ fractions from BMAs supplied from patients with MM were provided by the Blood Cancer Biobank Ireland.
Reagents and antibodies
Siglec-7 and Siglec-9 chimeras were from R&D Systems (1138-SL-050 and 1139-SL-050), anti-human immunoglobulin G Fc fragment was from Jackson ImmunoResearch (009-000-008). Maackia amurensis agglutinin was from Vector Laboratories (B-1265), 3Fax-Peracetyl Neu5Ac was from Merck (566224-10mg), daratumumab (Dara) (Janssen) was supplied by University Hospital Galway, and neuraminidase (Vibrio cholerae) was supplied by Roche (11 080 725 001). Carboxyfluorescein diacetate succinimidyl ester (CFSE) and cell fixation and permeabilization kit were from Thermo Fisher Scientific (C34554 and 00-5523-00). Tag-it dye was from Biolegend (425101). GolgiStop was from BD Biosciences (554724). Propidium iodide (PI) was from Sigma. (Antibody details are located in Supplementary Materials.)
Siglec ligand staining on MM cell lines and primary samples
MM cell lines or isolated primary MM cells were stained with Siglec-7 and Siglec-9 chimeras and then washed and stained with APC AffiniPure F(ab)2 fragment goat anti-human immunoglobulin G, Fc-γ fragment specific. Dead cells were excluded using PI staining.
Siglec-7 ligand pulldown and analysis using mass spectrometry
MM1S, JJN3, and H929 MM cell pellets were lysed, untreated or treated, with neuraminidase (NEURA). Lysates were then incubated with magnetic bead–Siglec-7 Fc chimera complexes, and bound ligands were subsequently isolated and analyzed using mass spectrometry. Full methodology for Siglec-7 ligand pulldown and mass spectrometry analysis is available in Supplementary Materials.
NK cell expansion
Fresh peripheral blood or buffy coats were obtained from healthy donors after informed consent. Primary NK cells were isolated by negative selection (Miltenyi) and expanded using NK cell expansion media (Miltenyi) supplemented with 5% heat inactivated human AB serum (Sigma) and 500 IU/mL of IL-2. Expansions were cultured at 37°C, 5% CO2.
MM cell desialylation assays
Briefly, JJN3, H929, MM1S, K562, and primary MM cells were pretreated with 0.1 U/mL of the sialidase NEURA or glycobuffer (GLYCO) control or treated with 300 µM of 3Fax-Peracetyl Neu5Ac (sialyltransferase inhibitor, SIA) or DMSO control for 72 hours and subsequently cocultured with CFSE-stained KHYG-1 or primary NK cells (naïve, IL-2 activated, and expanded) at indicated effector/target (E:T) ratios in 4-hour cytotoxicity assays in flat-bottomed 96-well plates. Total MM cell death (CFSE- cells) was determined by PI staining.
In cytotoxicity assays using Dara, JJN3 and H929 were treated with SIA as described previously and treated with 10 µg/mL of Dara for 30 minutes prior to being cocultured with specific NK cells. JJN3 were treated with 10 nM of ATRA in combination with 300 µM of SIA prior to treatment with Dara and cocultured with NK cells as described previously.
Percent specific lysis was calculated using the following formula:
NK cell degranulation assays
1 × 106 JJN3, H929, and K562 cells were treated with NEURA and then cultured with IL-2 activated (500 U/mL) NK cells at a 1:1 E:T ratio. After 1 hour, cells were collected and stained for CD56, CD3, Siglec-7, Siglec-9, and CD107a along with LIVE/DEAD stain (Invitrogen) before being analyzed or fixed using 4% paraformaldehyde.
NK cell intracellular cytokine staining assays
1 × 106 JJN3, H929, and K562 cells were treated with NEURA and then cocultured with Tag-it dye-stained expanded primary NK cells at a 1:1 E:T ratio. NK cells were stained with an anti-CD107a antibody at the onset of coculture, and Golgi-stop was added 1 hour into the assay. After a total of 5 hours of coculture, cells were collected, fixed, and permeabilized according to the supplier’s instructions (FIX/PERM kit; Thermo Fisher). Cells were then stained with anti–tumor necrosis factor (TNF) α and interferon (IFN) γ antibodies before being analyzed using flow cytometry. NK cells were determined as Tag-it+.
Siglec-7 gene editing using CRISPR/Cas9
Freshly isolated NK cells were cultured in expansion media for 6 to 8 days. Briefly, 1 ×106 NK cells were mixed with a gRNA-tracrRNA CRISPR ribonucleoprotein complex and Cas9 enzyme (5:1 molar ratio) (Synthego) targeting Siglec-7. The NK cell–RNP complex was electroporated using the MaxCyte GT transfection system and cultured for 6 to 8 days before analyzing efficiency of gene knockout (KO) using flow cytometry. Functional cytotoxicity assays were performed as described previously with mock-electroporated NK cells or Siglec-7-targeted NK cells on days 6 to 9.
Analysis of Siglec-7 and Siglec-9 expression on immune cell subsets from MM patient samples
CD138- fractions isolated from BMAs of patients with MM were stained using SYTOX Blue, anti-CD3, anti-CD56, anti-CD4, anti-CD8, anti-CD11b, and anti-CD14 antibodies, allowing the identification of different immune subsets. NK cells were determined as CD56+/CD3-, T cells were determined as CD56-/CD3+ and CD4+ or CD8+ for CD4 and CD8 T cells, respectively. Macrophages were determined as CD56-/CD3- and CD11b+/CD14+. Siglec-7 and Siglec-9 expression was measured. These data were compared with the expression of Siglec-7 and Siglec-9 on the same immune cell subsets from peripheral blood of healthy donors.
Data and statistics
Flow cytometry data were analyzed using FlowJo version 10 software. Results were analyzed using GraphPad Prism version 7.0. Comparisons between 2 groups were analyzed by using either a Mann-Whitney unpaired t-test or Student’s paired t-test. Comparisons between multiple groups were analyzed by repeated measure one-way analysis of variance (ANOVA). Data represent mean ± SD or SEM, where indicated (*P < .05; **P < .01; ***P < .001; ****P < .0001 and were considered statistically significant).
Ethics approval and consent to participate
Peripheral blood was sourced from informed, consenting healthy donors for the isolation of primary NK cells. Ethical approval is as follows: NUI Galway, C.A. 1805; Karolinska Institutet, Dnr 2006/229-31/3. BMAs were provided by informed consenting patients at University Hospital Galway. Ethical approval is as follows: NUI Galway, C.A. 662.
Results
Ligands for Siglec-7 and Siglec-9 and their cognate receptors are expressed by primary MM and NK cells, respectively
Ligands for Siglec-7 and Siglec-9 have previously been shown to be expressed by a multitude of cancerous cell types.18,19,23 -25 To determine the expression of these ligands in MM, we used recombinant Siglec-7 and Siglec-9-Fc chimeras to screen primary MM cells and MM cell lines. This analysis revealed high expression of both Siglec-7 and Siglec-9 ligands (Siglec-7L, Siglec-9L) on primary MM cells from both monoclonal gammopathy of undetermined significance (MGUS) and newly diagnosed patients and on MM cell lines MM1S, H929, and JJN3 (Figure 1A-B, respectively). By analyzing the mean fluorescence intensity (MFI) of Siglec-7L on Siglec-7L+ MM cells, we observed that MM1S had the highest expression of Siglec-7L followed by JJN3 and H929 (supplemental Figure 1A). In contrast, Siglec-9L expression on MM cells was at comparable levels among H929, MM1S, and JJN3 (supplemental Figure 1B).
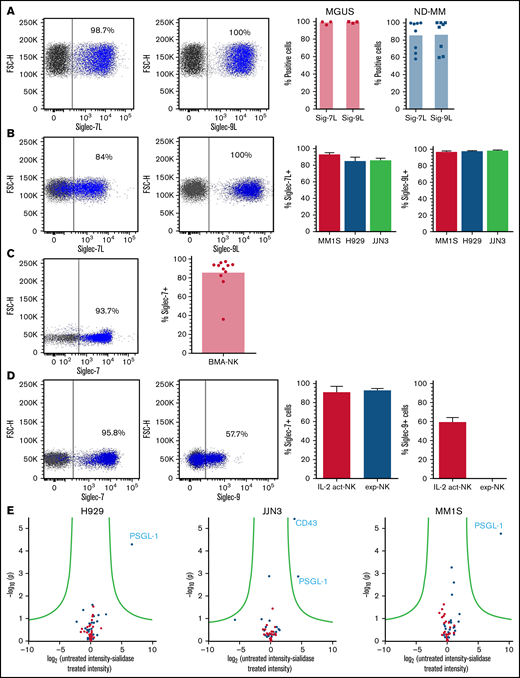
Siglec-7 and Siglec-9 ligands, and their concomitant receptors, are expressed by MM and NK cells, respectively. Using recombinant Siglec-7 and Siglec-9 chimeras, primary MM cells isolated from BMAs supplied by MGUS and newly diagnosed (ND) patients (A) and a panel of commonly used MM cell lines (B) were stained for Siglec-7L and Siglec-9L expression. (C) Using a fluorescently labeled anti–Siglec-7 antibody primary NK cells from BMAs of patients with MM were screened for the expression of Siglec-7. (D) Primary NK cells (IL-2 activated and expanded) were stained for Siglec-7 and Siglec-9 expression. (E) To elucidate the identity of Siglec-7L in MM, mass spectrometry was carried out on proteins bound to Siglec-7 Fc chimera-magnetic bead complexes after incubation with MM1S, H929, and JJN3 cell lysates untreated or treated with neuraminidase. (A-D) Combined data represented as bar graphs and an representative dot blot from one individual MM BMA sample or biological repeat. Data in E are represented as a volcano plot. n = 8 independent samples for ND. (A) n = 3 for MGUS. (B) n = 3 biological replicates. (C) n = 11; (D) n = 7; (E) n = 3.
Siglec-7 and Siglec-9 ligands, and their concomitant receptors, are expressed by MM and NK cells, respectively. Using recombinant Siglec-7 and Siglec-9 chimeras, primary MM cells isolated from BMAs supplied by MGUS and newly diagnosed (ND) patients (A) and a panel of commonly used MM cell lines (B) were stained for Siglec-7L and Siglec-9L expression. (C) Using a fluorescently labeled anti–Siglec-7 antibody primary NK cells from BMAs of patients with MM were screened for the expression of Siglec-7. (D) Primary NK cells (IL-2 activated and expanded) were stained for Siglec-7 and Siglec-9 expression. (E) To elucidate the identity of Siglec-7L in MM, mass spectrometry was carried out on proteins bound to Siglec-7 Fc chimera-magnetic bead complexes after incubation with MM1S, H929, and JJN3 cell lysates untreated or treated with neuraminidase. (A-D) Combined data represented as bar graphs and an representative dot blot from one individual MM BMA sample or biological repeat. Data in E are represented as a volcano plot. n = 8 independent samples for ND. (A) n = 3 for MGUS. (B) n = 3 biological replicates. (C) n = 11; (D) n = 7; (E) n = 3.
Next, we performed flow cytometry analyses of patient and healthy donor-derived NK cells and NK cell lines. This analysis showed strong expression of Siglec-7 on NK cells from patient BMAs (Figure 1C) as well as on healthy naïve, IL-2 activated and expanded NK cells (Figure 1D). Siglec-9 was expressed by a subset of IL-2–activated NK cells but absent in expanded NK cells (Figure 1D). Analysis of KHYG-1 and NK-92 NK cell lines revealed partial Siglec-7 expression whereas expression of Siglec-9 was absent (supplemental Figure 1C).
In MM there have been no previous attempts to elucidate the identity of Siglec-7 ligands. To address this knowledge gap, Siglec-7 ligands were immunoprecipitated from MM cell lysates (untreated or pretreated with NEURA) and identified using mass spectrometry. Cell lysates were prepared from Siglec-7L+ MM1S, H929, and JJN3 cell lines (Figure 1C). Immunoprecipitated cell surface and secreted proteins that were enriched from untreated vs sialidase-treated lysates represent sialic acid-dependent ligands for Siglec-7. P-selectin glycoprotein ligand-1 (PSGL-1) emerged as the primary Siglec-7L expressed in MM1S, H929, and JJN3 cell lines. Furthermore, CD43 was also identified as a Siglec-7L expressed in JJN3 cells, consistent with previous work showing that CD43 is a high affinity ligand for Siglec-7 (Figure 1E).21 These results identify PSGL-1 as a new, broadly expressed Siglec-7L in MM.
Finally, analysis of NK cells from BMAs derived from patients with MM revealed an increase in Siglec-7 expression compared with NK cells from the peripheral blood of healthy donors, as determined by comparing MFI of Siglec-7+ cells (Figure 2A). Further analysis revealed decreased Siglec-9 expression, as determined by both overall Siglec-9+ cells and MFI of Siglec-9+ cells, compared with healthy donor-derived controls (Figure 2B).
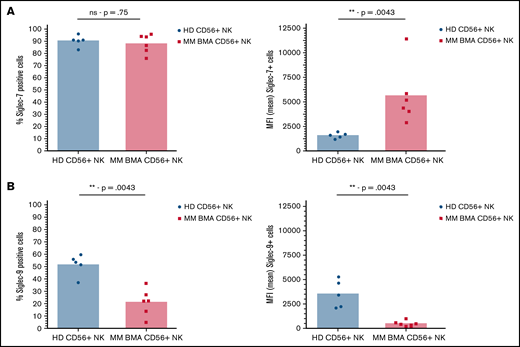
Siglec-7 expression is increased on NK cells from BMAs of patients with MM, while Siglec-9 expression is decreased. CD56+/CD3- NK cells sourced from peripheral blood of healthy donors (black) and from BMAs of patients with MM (red) were stained for the expression of (A) Siglec-7 and (B) Siglec-9. Data analyzed using Mann-Whitney unpaired t-test. Graphs represent both mean Siglec-7+ NK cells and mean MFI of Siglec-7+ NK cells as well as individual values for each sample. n = 5 for healthy donor peripheral blood-derived NK cells, and n = 6 for MM patient BMA-derived NK cells. **P < .01; ns, not significant.
Siglec-7 expression is increased on NK cells from BMAs of patients with MM, while Siglec-9 expression is decreased. CD56+/CD3- NK cells sourced from peripheral blood of healthy donors (black) and from BMAs of patients with MM (red) were stained for the expression of (A) Siglec-7 and (B) Siglec-9. Data analyzed using Mann-Whitney unpaired t-test. Graphs represent both mean Siglec-7+ NK cells and mean MFI of Siglec-7+ NK cells as well as individual values for each sample. n = 5 for healthy donor peripheral blood-derived NK cells, and n = 6 for MM patient BMA-derived NK cells. **P < .01; ns, not significant.
Targeted desialylation of MM cells potentiates NK cell activation and tumor cell lysis
To investigate the potential role of hypersialylation in regulating NK cell cytotoxicity, MM cells were selectively desialylated using either NEURA or SIA.
NEURA treatment resulted in a near complete abolishment of Siglec-7L and Siglec-9L expression on K562, H929, and JJN3 cells (supplemental Figure 2A). Additionally, significantly enhanced NK cell–mediated lysis of desialylated MM cells was observed by naive, IL-2 activated, and expanded NK cells (Figure 3A-C). Additionally, enhanced KHYG-1-mediated cytotoxicity was observed in cocultures with NEURA-treated JJN3 (supplemental Figure 2B). NEURA treatment was observed to be toxic to MM1S (supplemental Figure 2C). Thus, a need for a gentler approach to desialylate MM cells was required. Using the cell-permeable sialic acid analog SIA, sialyltransferase activity was inhibited in MM1S cells without toxicity, and enhanced expanded NK cell–mediated lysis of MM cells upon desialylation by SIA was observed (supplemental Figure 2D).
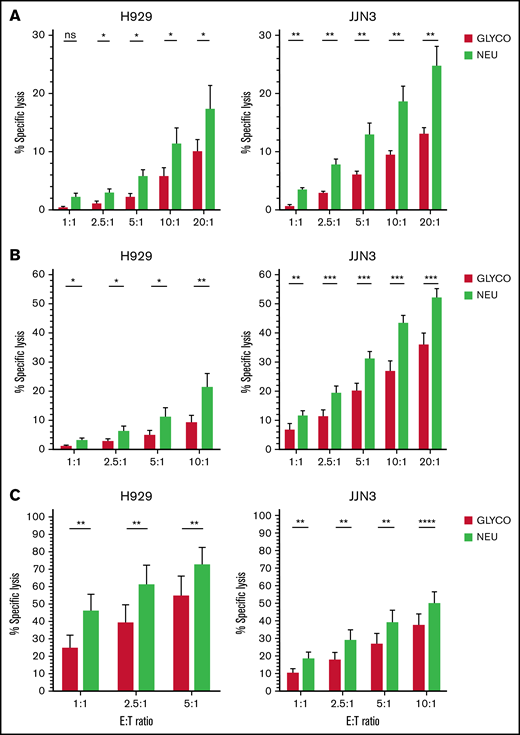
Desialylation of the MM cell surface enhances NK cell–mediated tumor cell lysis. (A-C) JJN3 and H929 MM cell lines were treated with neuraminidase (NEURA) or glycobuffer (GLYCO) and cocultured with healthy donor peripheral blood-derived NK cells that were naïve (A), IL-2 activated overnight (B), and expanded at various indicated E:T ratios (C). All cytox assays were carried out for 4 hours. Graphs represent mean specific lysis + SEM. (A-C) n = 7. Data analyzed using Student’s paired t-test. *P < .05; **P < .01; ***P < .001; ****P < .0001; ns, not significant.
Desialylation of the MM cell surface enhances NK cell–mediated tumor cell lysis. (A-C) JJN3 and H929 MM cell lines were treated with neuraminidase (NEURA) or glycobuffer (GLYCO) and cocultured with healthy donor peripheral blood-derived NK cells that were naïve (A), IL-2 activated overnight (B), and expanded at various indicated E:T ratios (C). All cytox assays were carried out for 4 hours. Graphs represent mean specific lysis + SEM. (A-C) n = 7. Data analyzed using Student’s paired t-test. *P < .05; **P < .01; ***P < .001; ****P < .0001; ns, not significant.
We next confirmed enhanced NK cell activation when cocultured with desialylated target cells. IL-2–activated NK cells cocultured with NEURA-treated MM cells had increased levels of degranulation, determined by expression of cell surface CD107a, compared with cocultures with GLYCO-treated controls (Figure 4A-B). Analysis of NK cell subsets expressing or lacking Siglec-7 and/or Siglec-9 revealed a more prominent increase in degranulation by Siglec-7+ expressing NK cells compared with Siglec-7- NK cells (Figure 4C). Interestingly, there was also a minor increase in degranulation by Siglec-7-/Siglec-9- NK cells. Similar to IL-2–activated NK cells, expanded NK cells had elevated levels of CD107a expression when cocultured with desialylated K562, H929, and JJN3 (Figure 4D). Finally, expanded NK cells had increased levels of TNF-α and IFN-γ when cocultured with desialylated K562, whereas similar nonsignificant trends were observed against desialylated H929 and JJN3 (Figure 4E).
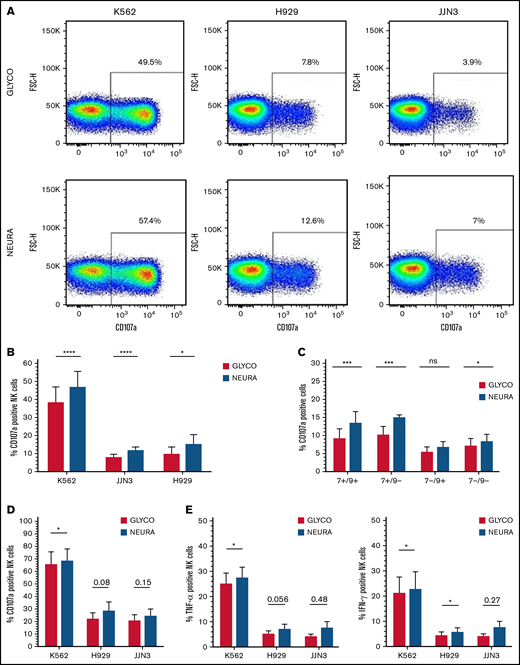
Desialylation of MM cells increases NK cell degranulation and surface-expressed CD107a after coculture. (A) IL-2 activated NK cells were cocultured with K562, JJN3, or H929 ± desialylation using NEURA or GLYCO for 1 hour, after which cells were collected and CD107a expression was measured on bulk NK cells. Histogram representative of n = 1 biological repeat for each cell under each condition. (B) CD107a expression on IL-2 activated primary NK cells exposed to NEURA-treated K562, JJN3, and H929 was determined and compared with CD107a expression on NK cells cocultured with GLYCO-treated controls. (C) NK cells were subdivided based on Siglec-7/Siglec-9 expression and subset degranulation were measured after coculture with JJN3 treated with either GLYCO or NEURA. (D) CD107a expression was measured on expanded primary NK cells exposed to NEURA-treated K562, JJN3, and H929 and compared with CD107a expression on NK cells cocultured with GLYCO-treated controls. (E) TNF-α and IFN-γ expression within NK cells was measured after coculture with NEURA-treated K562, JJN3, and H929 and compared with the expression of TNF-α and IFN-γ when cocultured with GLYCO-treated controls. (B-E) Data analyzed using Student’s paired t-test; graphs represent mean CD107α/TNF-α/IFN-γ positive NK cells +SEM. An individual repeat of n = 7 donors (A), n = 7 (B-C), n = 5 (D-E). *P < .01; ***P < .001; ****P < .0001; ns, not significant.
Desialylation of MM cells increases NK cell degranulation and surface-expressed CD107a after coculture. (A) IL-2 activated NK cells were cocultured with K562, JJN3, or H929 ± desialylation using NEURA or GLYCO for 1 hour, after which cells were collected and CD107a expression was measured on bulk NK cells. Histogram representative of n = 1 biological repeat for each cell under each condition. (B) CD107a expression on IL-2 activated primary NK cells exposed to NEURA-treated K562, JJN3, and H929 was determined and compared with CD107a expression on NK cells cocultured with GLYCO-treated controls. (C) NK cells were subdivided based on Siglec-7/Siglec-9 expression and subset degranulation were measured after coculture with JJN3 treated with either GLYCO or NEURA. (D) CD107a expression was measured on expanded primary NK cells exposed to NEURA-treated K562, JJN3, and H929 and compared with CD107a expression on NK cells cocultured with GLYCO-treated controls. (E) TNF-α and IFN-γ expression within NK cells was measured after coculture with NEURA-treated K562, JJN3, and H929 and compared with the expression of TNF-α and IFN-γ when cocultured with GLYCO-treated controls. (B-E) Data analyzed using Student’s paired t-test; graphs represent mean CD107α/TNF-α/IFN-γ positive NK cells +SEM. An individual repeat of n = 7 donors (A), n = 7 (B-C), n = 5 (D-E). *P < .01; ***P < .001; ****P < .0001; ns, not significant.
Desialylation of the MM cell surface does not unmask activating ligands for NKG2D
It has been hypothesized that hypersialylation can mask activating ligands, typically expressed by malignantly transformed cells, for the activating NK cell receptor natural killer group 2 member D (NKG2D).6 To address this, we desialylated K562, MM1S, JJN3, and H929 cell lines using NEURA and compared the expression of the NKG2D ligands MICA/B and UL16-binding protein (ULBP) 1-6 to that of GLYCO-treated controls. This revealed no increase in expression of MICA/B and ULBP-1-6 in any of the MM cell lines. However, increased expression of ULPB-2/5/6 and ULPB-3 was observed on the erythroleukemia cell line K562 (supplemental Figure 3).
MM cell desialylation results in enhanced CD38 detection that can be used to enhance NK cell–mediated ADCC of MM cells
Previously, desialylation of MM cell lines has been shown to result in an increase in detectible B-cell maturation antigen, a target antigen for monoclonal antibody (moAb)-based therapies.26 To further investigate this, the expression of several antigens targeted by therapeutic moAbs was compared upon desialylation to expression of the same antigens on GLYCO-treated controls.
After desialylation, a consistent and reproducible increase in the MFI of CD38 on CD38+ cells was observed on MM1S and H929 cell lines but not in JJN3 (Figure 5A-B). Finally, MNCs were isolated from BMAs supplied by patients with MM and treated with SIA for 60 hours. Similar to MM1S and H929, increases in CD38 MFI were observed on CD38+/CD138+ MM cells (Figure 5C).
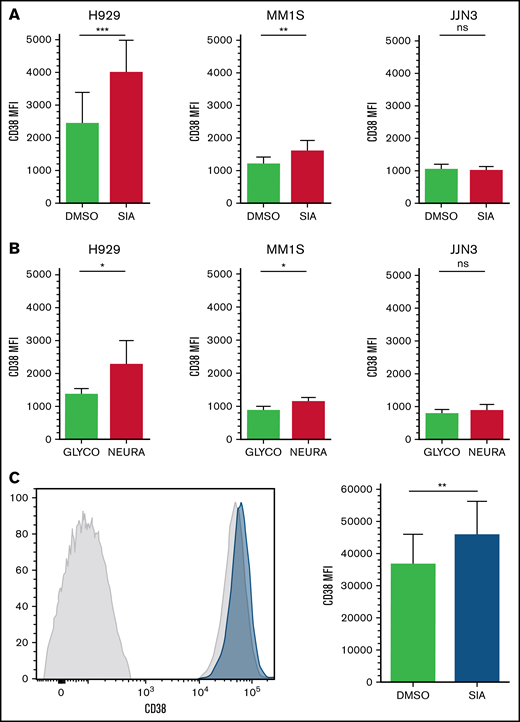
CD38 expression is increased on MM and NK cells after cell surface desialylation. (A-B) MM cell lines H929, MM1S, and JJN3 were pretreated with either NEURA (A) or SIA (B) and stained for CD38 expression. (C) MNCs isolated from BMAs of afflicted patients were treated with SIA for 60 hours, after which CD38 expression was measured. n = 5 biological replicates (A-B), n = 6 primary samples (C). Data analyzed using Student’s paired t-test; graphs represent mean CD38 mean fluorescence intensity (MFI) + SEM. *P < .05; **P < .01; ***P < .001; ns, not significant.
CD38 expression is increased on MM and NK cells after cell surface desialylation. (A-B) MM cell lines H929, MM1S, and JJN3 were pretreated with either NEURA (A) or SIA (B) and stained for CD38 expression. (C) MNCs isolated from BMAs of afflicted patients were treated with SIA for 60 hours, after which CD38 expression was measured. n = 5 biological replicates (A-B), n = 6 primary samples (C). Data analyzed using Student’s paired t-test; graphs represent mean CD38 mean fluorescence intensity (MFI) + SEM. *P < .05; **P < .01; ***P < .001; ns, not significant.
One of the key mechanisms of action of the anti-CD38 moAb Dara in MM is to trigger NK cell–mediated antibody-dependent cellular-mediated cytotoxicity (ADCC) of tumor cells.27 Siglec-7 has previously been implicated in reducing the efficacy of trastuzumab-induced ADCC in breast cancer.28 Thus, we next decided to determine the role of Siglec-7 in regulating the efficacy of Dara-induced ADCC in MM. Having observed enhanced MM cell surface CD38 expression upon desialylation, we combined desialylation with Dara treatment to maximize NK cell anti-MM cytotoxicity.
H929 and JJN3 were pretreated with SIA prior to exposure to Dara in cocultures with primary expanded NK cells. H929 treated with both SIA and Dara were more readily lysed by primary expanded NK cells than H929 treated with either SIA or Dara alone (Figure 6A). In contrast, the levels of NK cell–mediated lysis of JJN3 treated with SIA and Dara were comparable to JJN3 treated with SIA alone (Figure 6A).
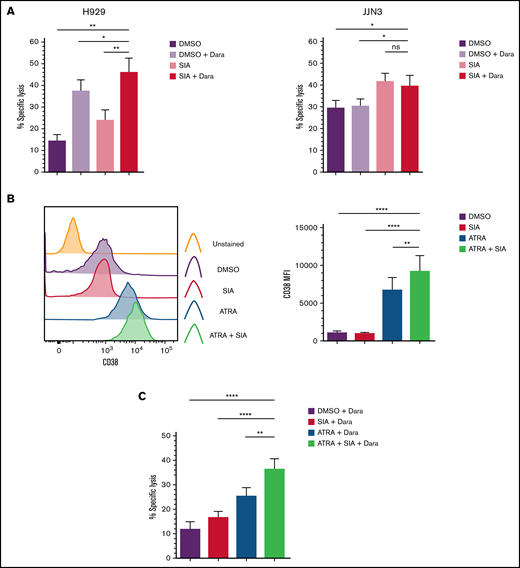
Desialylation in combination with Daratumumab treatment enhances NK cell–mediated clearance of MM cells. (A) H929 and JJN3 were treated with DMSO alone (black), DMSO + Dara (gray), SIA alone (orange), and SIA + Dara (red) and were subsequently cocultured with expanded primary NK cells at a 1:1 E:T ratio. (B) JJN3 MM cells were treated with SIA alone (black), all-trans retinoic acid (ATRA) alone (blue), and SIA + ATRA (green) for 72 hours, after which CD38 expression was measured. (C) JJN3 were treated with DMSO alone (black), SIA alone (red), ATRA alone (blue), and SIA + ATRA (green) for 72 hours prior to Dara treatment and coculture with expanded primary NK cells at a 1:1 E:T ratio. Bar graphs represent mean specific lysis + SEM or CD38 MFI + SEM. Data analyzed using one-way ANOVA. n = 5 (A-C); n = 6 (B). *P < .05; **P < .01; ****P < .0001; ns, not significant.
Desialylation in combination with Daratumumab treatment enhances NK cell–mediated clearance of MM cells. (A) H929 and JJN3 were treated with DMSO alone (black), DMSO + Dara (gray), SIA alone (orange), and SIA + Dara (red) and were subsequently cocultured with expanded primary NK cells at a 1:1 E:T ratio. (B) JJN3 MM cells were treated with SIA alone (black), all-trans retinoic acid (ATRA) alone (blue), and SIA + ATRA (green) for 72 hours, after which CD38 expression was measured. (C) JJN3 were treated with DMSO alone (black), SIA alone (red), ATRA alone (blue), and SIA + ATRA (green) for 72 hours prior to Dara treatment and coculture with expanded primary NK cells at a 1:1 E:T ratio. Bar graphs represent mean specific lysis + SEM or CD38 MFI + SEM. Data analyzed using one-way ANOVA. n = 5 (A-C); n = 6 (B). *P < .05; **P < .01; ****P < .0001; ns, not significant.
Based on these data, we confirm that hypersialylation masks expression of CD38 on MM cells, and detection of CD38 is increased upon desialylation. Furthermore, we conclude that desialylation can be combined with the anti-CD38 moAb Dara to enhance NK cell–mediated ADCC of CD38-expressing MM cells.
ATRA treatment enhances CD38 expression, and combination treatment with SIA and daratumumab strongly enhances NK cell–mediated cytotoxicity
Because the MFI of CD38 on JJN3 cells was not increased upon desialylation, we next decided to upregulate CD38 on JJN3. All-trans-retinoic acid (ATRA) is used to upregulate CD38 on myeloid cells.29 Previously, ATRA treatment of both primary MM cells and MM cell lines upregulated CD38 expression.30 We therefore decided to upregulate CD38 on JJN3 using ATRA, and combine ATRA treatment with SIA to investigate whether a further exposure of CD38 could be obtained.
As shown in Figure 6B, a >6-fold increase in CD38 MFI was observed on JJN3 treated with ATRA, and a >8-fold increase of CD38 MFI was observed with both ATRA and SIA (Figure 6B). Considering this, we next cocultured JJN3 cells that had been exposed to SIA and ATRA, or the respective compounds alone, with Dara and expanded NK cells. JJN3 treated with SIA, ATRA, and Dara were more susceptible to NK cell–mediated lysis than ATRA and Dara or SIA- and Dara-treated cells (Figure 6C). This analysis also revealed that JJN3 treated with ATRA and Dara were more readily targeted than JJN3 treated with either DMSO and Dara or SIA and Dara.
Genetic deletion of Siglec-7 augments NK cell cytotoxicity against Siglec-7L+/CD38+ MM cells
To further characterize the role of the Siglec-Siglec ligand axis in NK cell–mediated targeting of MM, we next decided to address this following interference with Siglec-7 expression. While blockade of Siglec-7 and Siglec-9 using moAbs has previously been shown to enhance NK cell cytotoxicity against cancerous cell lines, genetically modifying NK cells to abolish Siglec expression had not been attempted.
We disrupted Siglec-7 in expanded NK cells using the CRISPR-Cas9 gene editing technology. After transfection of expanded NK cells with a gRNA-Cas9 RNP complex, 91% ± 3.8% of the cells lost cell surface expression of Siglec-7 (Siglec-7KO) as compared with mock electroporated (“mock”) NK cells, 6 to 8 days after transfection (Figure 7A-B). We next cocultured both mock or Siglec-7KO NK cells with Siglec-7L+ MM cell lines JJN3 and H929 to address the functional consequence of NK cells lacking Siglec-7 expression. This analysis revealed enhanced killing of JJN3 and H929 by Siglec-7KO NK cells as compared with mock NK cells (Figure 7C-D). Variations in enhancement of cytotoxicity were observed between donors within the range of 5% to 25%, corresponding to 1.3- to 1.7-fold increases compared with Siglec-7+/+ NK cells.
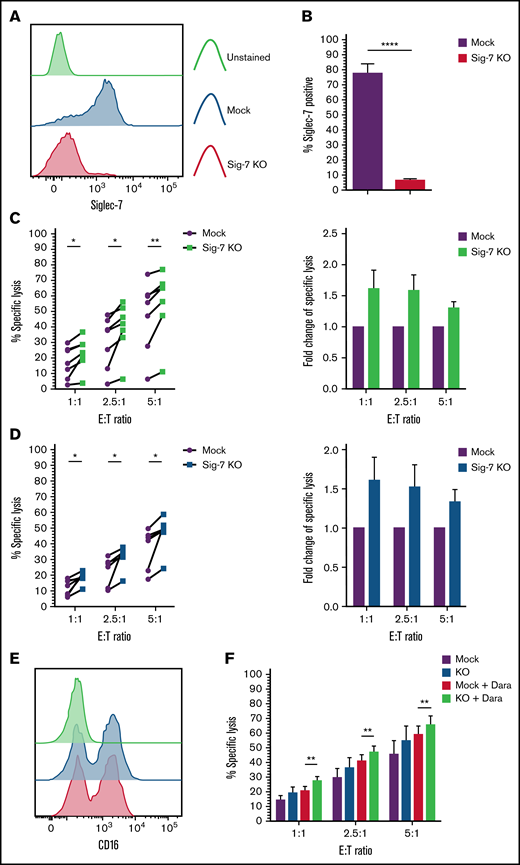
Targeted knockout of Siglec-7 using CRISPR/Cas9 enhances NK cell–mediated cytotoxicity toward Siglec-7L+/CD38+ MM cell lines. Siglec-7 was targeted for KO using the CRISPR/Cas9 system and the MaxCyte GT transfection system. NK cells were isolated and expanded from peripheral blood of healthy donors for 10 to 14 days prior to transfection. Cytotoxicity assays using CRISPR/Cas9-targeted NK cell were carried out 6 to 9 days after transfection. Siglec-7 KO was determined using flow cytometry, and efficiency was analyzed and displayed as a histogram (A) representative of n = 1 Siglec-7 KO readout on viable NK cells and bar graphs (B) of mock electroporated and CRISPR-targeted NK cells representing complete KO of Siglec-7 in n = 7 donors. Functionality of Siglec-7 KO NK cells was measured in cytotoxicity assays against the Siglec-7L+ MM cell lines H929 (C) and JJN3 (D). (E) CD16 expression was recorded on Mock and Siglec-7KO NK cells using flow cytometry. (F) Mock and Siglec-7KO NK cells were cocultured with Dara-treated CD38+ MM1S MM cells in cytotoxicity assays. Cytox assays were carried out for 4 hours; graphs represent mean specific lysis + SEM. n = 7 biological replicates (B-C); n = 6 (F). (B-D) Data analyzed using Student’s paired t-test; (F) data analyzed using one-way ANOVA. (C-D) Data represent individual values recorded in n = 7 biological replicates and fold change in specific lysis by both mock and Siglec-7KO NK cells. *P < .05; **P < .01; ****P < .0001.
Targeted knockout of Siglec-7 using CRISPR/Cas9 enhances NK cell–mediated cytotoxicity toward Siglec-7L+/CD38+ MM cell lines. Siglec-7 was targeted for KO using the CRISPR/Cas9 system and the MaxCyte GT transfection system. NK cells were isolated and expanded from peripheral blood of healthy donors for 10 to 14 days prior to transfection. Cytotoxicity assays using CRISPR/Cas9-targeted NK cell were carried out 6 to 9 days after transfection. Siglec-7 KO was determined using flow cytometry, and efficiency was analyzed and displayed as a histogram (A) representative of n = 1 Siglec-7 KO readout on viable NK cells and bar graphs (B) of mock electroporated and CRISPR-targeted NK cells representing complete KO of Siglec-7 in n = 7 donors. Functionality of Siglec-7 KO NK cells was measured in cytotoxicity assays against the Siglec-7L+ MM cell lines H929 (C) and JJN3 (D). (E) CD16 expression was recorded on Mock and Siglec-7KO NK cells using flow cytometry. (F) Mock and Siglec-7KO NK cells were cocultured with Dara-treated CD38+ MM1S MM cells in cytotoxicity assays. Cytox assays were carried out for 4 hours; graphs represent mean specific lysis + SEM. n = 7 biological replicates (B-C); n = 6 (F). (B-D) Data analyzed using Student’s paired t-test; (F) data analyzed using one-way ANOVA. (C-D) Data represent individual values recorded in n = 7 biological replicates and fold change in specific lysis by both mock and Siglec-7KO NK cells. *P < .05; **P < .01; ****P < .0001.
Thus, we demonstrate for the first time that primary expanded NK cells can be genetically modified to KO Siglec-7 using CRISPR/Cas9, resulting in enhanced cytotoxicity against Siglec-7L+ MM cells.
Deletion of Siglec-7 in combination with daratumumab treatment of MM cells is more effective at tumor cell lysis than either approach alone
Adoptive transfer of genetically modified NK cells poses an exciting novel alternative for cellular therapies.31 Thus, we decided to combine Siglec-7KO with Dara treatment to ascertain whether Siglec-7KO NK cells would retain their capability to carry out ADCC. Confirmatory flow cytometry staining showed that Siglec-7KO did not affect CD16 expression on NK cells (Figure 7E). Cytotoxicity assays revealed that Siglec-7KO NK cells cocultured with MM1S pretreated with Dara resulted in enhanced NK cell cytotoxicity compared with Mock NK cells cocultured with MM1S pre-treated with Dara (Figure 7F).
Discussion
We set out to establish whether hypersialylation plays a role in facilitating immune evasion of NK cells in MM and identify potential therapeutic strategies to abrogate this problem and NK cell anticancer functions.
We first confirmed the potential for the Siglec-Siglec ligand axis to be hijacked by MM cells by observing Siglec ligand and receptor expression on MM cell and NK cells, respectively. Comparable expression of both Siglec-7L and Siglec-9L was observed on CD138+ plasma cells from patients with monoclonal gammopathy of undetermined significance (MGUS). Because we were not able to evaluate Siglec ligand expression on normal plasma cells, we cannot conclude that malignant plasma cells have higher levels in the resting state. However, while Siglec-7/9L ligand expression is not solely restricted to cancer cells, it has been observed that upon encountering NK cells, Siglec-7L and Siglec-9L expression on malignantly transformed cells is upregulated, increasing the inhibitory stimulus received by the NK cell.32
The identification of PSGL-1 as a prominent ligand for Siglec-7 in MM is important. PSGL-1 is highly expressed in MM biopsies as well as on MM cell lines.33 Interactions between PSGL-1 and P- and E-selectin regulate MM cell proliferation and homing and contribute to resistance to therapies.33-35 Indeed, antibody blockade of PSGL-1 increased retention of MM cells in the circulation, sensitizing them to the proteosome inhibitor bortezomib.34 Coupled with the elucidation of its role as a ligand for Siglec-7, PSGL-1 therefore represents an important target that could be used to enhance future NK cell–based adoptive therapies. The finding that CD43 also acts as a ligand for Siglec-7 in MM is not surprising given its recent recognition as an important Siglec-7 ligand in acute myeloid leukemia.21 However, these data suggest that PSGL-1 is likely to be the most prominent Siglec-7L on MM cells, representing a potential therapeutic target.
Desialylation of MM cells strongly enhanced primary NK cell–mediated cytotoxicity and degranulation, consistent with previous reports implicating hypersialylation in facilitating immune evasion.18,28,36 Increased degranulation was observed predominantly in Siglec-7–expressing NK cells, suggesting that Siglec-7 may be more influential than Siglec-9 in MM. Increased expression of IFN-γ and TNF-α in NK cells exposed to desialylated tumor cells indicates that desialylation of tumor cells may induce a more beneficial immunotherapeutic response than solely increasing NK cell–mediated cytotoxicity.
Increased detection of CD38 was observed on MM cells upon desialylation, which could have contributed to the higher levels of NK cell–mediated cytotoxicity observed in the presence of Dara when MM cells were pretreated with SIA. Because ATRA is a recognized means of inducing CD38 expression at a transcriptional level, we hypothesized that there could be a potential synergistic effect on CD38 expression when combined with SIA.30 Indeed, in CD38low JJN3, combined treatment with ATRA and SIA successfully restored CD38 expression, greatly enhancing Dara-induced ADCC. Because we observed a significant increase in CD38 on primary MM cells treated with SIA, this suggests that if a clinically viable means of desialylation were available, this dual strategy could be used in patients with MM to enhance the efficacy of CD38-targeting agents. One of the limitations of using Dara is self-targeting by NK cells in a process known as fratricide.37 To overcome this, CD38 has previously been deleted on expanded NK cells using CRISPR/Cas9, and CD38KO cells were demonstrated to be completely resistant to Dara-induced fratricide.37 Therefore, adoptive transfer of CD38KO NK cells, further edited to eliminate Siglec-7 or Siglec-9 expression or in combination with a desialylating agent, could elicit a potent anti-MM response.
Siglec-7 was expressed at a particularly high level on ex vivo expanded NK cells, indicating this to be a potential regulator of therapies involving the adoptive transfer of expanded NK cells.38 While we observed consistent increases in NK cell cytotoxicity by Siglec-7KO NK cells compared with mock electroporated controls, the magnitude of increase varied significantly between donors. The precise balance of activating/inhibitory receptor expression differs among individuals and in relation to a given target cell, indicating that Siglec-7KO could be more beneficial in patients with abnormally high Siglec-7 and/or Siglec-7L.39 However, given the expression of Siglec-9L on MM cells, we cannot exclude a role for Siglec-9 in evasion of NK cells by MM. Hypofunctional NK cell activity within the MM tumor microenvironment has been documented, and the multitude of factors, including hypoxia, cell-cell interactions, and dysregulated cytokine levels, all need to be considering when discussing how effective genetically modified NK cells may be against MM.40
Beyond Siglec-Siglec ligand interactions, we wished to explore whether sialylation could influence NK cell responsiveness through alternate mechanisms. While KO of Siglec-7 enhanced NK cell cytotoxicity against MM cells, in comparison with desialylating agents such as NEURA or SIA, the levels of increase in NK cell–mediated specific lysis observed were not as stark. Hypersialylation has been hypothesized to mask NKG2D ligands MICA/B and ULBP1-6, typically expressed by malignantly transformed cells.6,41 Furthermore, desialylation of breast cancer cells enhanced binding to recombinant NKG2D Fc chimeras.28 We did not observe any increase in MICA/B or ULBP1-6 expression upon desialylation of our panel of MM cell lines. It is worth noting that the erythroleukemia cell line K562 was used as a control for several of our experiments and, upon desialylation, a reproducible increase in the detection of ULBP-2/5/6 and ULBP-3 on K562 was observed. Therefore, there is a possibility for desialylation to confer a more potent NK cell anticancer response than individual targeting of Siglec-7, but this may depend on masking of NKG2D ligands by hypersialylation, or indeed their presence alone, on the target cancer type. Future studies to fully document the phenotype of ligands for NK cell receptors expressed by cancerous cells upon desialylation will likely help to identify the pathways mediating enhanced NK cell anticancer cytotoxicity.
Because we had already shown that the Siglec-7-sialic acid axis can be targeted by either desialylation or targeted deletion of Siglec-7 on NK cells, we aimed to show that Siglec-7KO NK cells could mediate enhanced ADCC induced by Dara. Indeed, Siglec-7KO NK cells induced a more robust cytotoxic response than control NK cells when combined with Dara. Therefore, genetic modification of NK cells using CRISPR/Cas9 to target checkpoint inhibitors represents a novel therapeutic approach by which NK cell cytotoxicity against cancer can be enhanced while still maintaining the NK cell’s ability to respond to moAb therapies.
Could these findings be translated to the clinic? Potential therapeutic strategies to overcome sialylation-induced immune evasion include the use of blocking antibodies, targeted delivery of sialidases-antibody conjugates, and sialyltransferase inhibitors. Previously, intratumoral injection of a sialyltransferase inhibitor in a syngeneic murine melanoma model was able to reverse tumor cell sialylation in vivo.42 After intratumoral injection of the inhibitor, a significant increase in NK cells and CD4+ and CD8+ T cells along with a significant depletion of regulatory T cells and myeloid cells was observed, suppressing tumor growth. The main challenge with the use of sialyltransferase inhibition is on target, off-tumor toxicity in the kidney, which necessitates targeted delivery to reduce systemic exposure and risk of nephrotoxicity.43,44 Sialidase-conjugated antibodies are another promising approach.28 Improved in vivo tumor control has been demonstrated when using sialidase-conjugated trastuzumab compared with unconjugated trastuzumab against a HER2+ expressing tumor in a syngeneic model.45
In conclusion, hypersialylation of tumor cells contributes to immune evasion of NK cells in MM. Desialylation, using either a sialidase or sialyltransferase inhibitor, is an effective strategy to abolish sialic acids on MM cells and enhance NK cell–mediated cytotoxicity. Additionally, desialylation can uncover CD38 expression and maximize NK cell–mediated ADCC in the presence of CD38 moAbs as well as disrupt the inhibitory Siglec-7-Siglec-7L axis, enhancing clearance of MM cells by NK cells. Alternatively, targeted deletion of the inhibitory Siglec-7 receptor using CRISPR/Cas9 enhances NK cell–mediated cytotoxicity. Therefore, targeted desialylation and/or Siglec-7 deletion may enhance the efficacy of adoptive NK cell immunotherapy in MM.
Acknowledgments
The authors would like to thank the Flow Cytometry Core Facility at NUI Galway, Ireland, and the Blood Cancer Biobank Ireland as well as the Flow Cytometry Core Facility at HERM, Karolinska Institutet, Sweden, for their help throughout this project.
This project was funded by biomedical research scholarship CRSDAL17 from the Irish Cancer Society and by Cancerfonden (Sweden); National Cancer Institute, National Institutes of Health, Postdoctoral Fellowship 1F32CA250324-01 and American Cancer Society Postdoctoral Fellowship PF-20-143-01-LIB (J.C.S.); N.M.R. acknowledges support from National Cancer Institute, National Institutes of Health Predoctoral to Postdoctoral Transition Award K00CA212454 (N.M.R.); and National Cancer Institute, National Institutes of Health (grant #CA227942) (C.R.B.).
Authorship
Contribution: J.D., M.C., and M.E.O.D. contributed equally to the work and wrote and reviewed themanuscript; S.S. and A.N. kindly helped to establish flow cytometry staining protocols and reviewed the manuscript; and J.C.S., N.M.R. and C.R.B. kindly carried out proteomics experiments and reviewed the manuscript.
Conflict-of-interest disclosure: C.R.B. is a co-founder and Scientific Advisory Board member of Lycia Therapeutics, Palleon Pharmaceuticals, Enable Bioscience, OliLux Bio, Grace Science LLC, Redwood Biosciences (a subsidiary of Catalent), and InterVenn Biosciences. The remaining authors declare no competing financial interests.
Correspondence: Michael E. O’Dwyer, National University of Ireland, G011 Biosciences, NUI Galway, Upper Newcastle, Galway, Ireland; e-mail: michael.odwyer@nuigalway.ie.

