

Key Points
HCK facilitates TLR/BCR crosstalk through activation of SYK in response to mutated MYD88.
The HCK inhibitor A419259 selectively blocks SYK activation in MYD88 mutated cell lines and primary WM lymphoplasmacytic cells.

Abstract
The SRC family kinase (SFK) HCK is transcriptionally upregulated and activated by mutated MYD88 (MYD88Mut), a key adaptor for Toll-receptor signaling. HCK activates BTK, AKT, and ERK in MYD88Mut lymphomas. SYK, a B-cell receptor (BCR) component, is activated in MYD88Mut lymphoma cells. Although the SFK LYN serves as a trigger for SYK activation in MYD88Mut ABC DLBCL cells, LYN activity is muted in MYD88Mut Waldenstrom macroglobulinemia (WM) cells. We therefore investigated a role for HCK in mediating SYK activation. Overexpression of wild-type (WT) (HCKWT) or gatekeeper mutated (HCKThr333Met) HCK in MYD88Mut lymphoma cells triggered SYK activation. Conversely, HCK knockdown reduced p-SYK in MYD88Mut lymphoma cells. Coimmunoprecipitation experiments showed that HCK was complexed with p-SYK in MYD88Mut BCWM.1 and TMD8 cells, but not in MYD88 WT Ramos cells. Rescue experiments in MYD88Mut lymphoma cells expressing HCKThr333Met led to persistent HCK and SYK activation and resistance to the HCK inhibitor A419259. Treatment of primary MYD88Mut WM cells with A419259 reduced p-HCK and p-SYK expression. Taken together, our findings show that SYK is activated by HCK in MYD88Mut B-cell lymphomas cells, broaden the prosurvival signaling generated by aberrant HCK expression in response to MYD88Mut, and help define HCK as an important therapeutic target in MYD88Mut B-cell lymphomas.
Introduction
Activating MYD88 mutations are common in B-cell malignancies, including Waldenstrom macroglobulinemia (WM) and activated B-cell diffuse large B-cell lymphoma (ABC DLBCL). MYD88 is a component of the Toll-like receptor (TLR) pathway. Mutated MYD88 (MYD88Mut) triggers assembly of a “Myddosome” complex leading to downstream prosurvival signaling that includes IRAK4/IRAK1-triggered NF-κB and HCK-mediated BTK/NF-κB, PI3K/AKT, and MAPK/ERK signaling.1-5 HCK is upregulated in response to mutated MYD88 signaling that includes STAT3, NF-κB, and the AP-1 complex component JunB in the presence of PAX5.6
The activation of the B-cell receptor (BCR) signaling component SYK has also been observed in MYD88Mut WM.7 Knockdown of MYD88 or a MYD88 signaling inhibitor abrogated SYK activation, whereas overexpression of mutated but not wild-type (WT) MYD88 amplified SYK activation in MYD88Mut and MYD88WT lymphoma cells.8 Importantly, knockdown of SYK or SYK inhibitors blocked p-STAT3 and p-AKT signaling and decreased viability of MYD88Mut lymphoma cells.8 In ABC DLBCL, chronic active BCR signaling underlies SYK activation, and knockdown of SYK decreases cell viability, including those harboring MYD88 and CD79 mutations.9 Although a role for the SRC family kinase (SFK) LYN has been proposed to trigger BCR/SYK activation in ABC DLBCL, other findings suggest an inhibitory role through BCR downmodulation.9-11 Herein, we investigated if HCK, an SFK that is normally downregulated in late-stage B-cell ontogeny, and transcriptionally upregulated and activated by MYD88Mut could trigger SYK activation, and thereby facilitate TLR/BCR crosstalk.4,11,12
Methods
Cell lines and drug treatment
MYD88Mut (MYD88Leu265Pro) WM (BCWM.1 and MWCL-1) and ABC DLBCL (TMD-8, HBL1, OCI-Ly3) cells; MYD88Mut (MYD88Ser222Arg) ABC DLBCL (SU-DHL-2) cells; and MYD88 WT (MYD88WT) GCB DLBCL (OCI-Ly7, OCI-Ly19), Burkitt lymphoma (Ramos) and myeloma (RPMI-8226) cells were used. Cell line identities were confirmed by GenePrint (Promega, WI). Ibrutinib and A419259 were obtained from MedChem Express (Monmouth Junction, NJ).
Patient samples and drug treatment
Bone marrow mononuclear cells were isolated as before, and 2 × 106 bone marrow mononuclear cells were treated for 1 to 2 hours with ibrutinib or A419259.4 PhosFlow analyses were performed on CD20+ lymphoplasmacytic lymphoma cells (LPCs), and western blotting was performed on CD19+ LPCs as before.3,4 MYD88 genotyping was performed by allele-specific polymerase chain reaction.13 CXCR4 mutation status was determined by allele-specific polymerase chain reaction and Sanger sequencing.14 Healthy donor CD19+ B cells from peripheral blood mononuclear cells were used as controls for LYN and p-LYN assessments. Sample use was approved by Dana-Farber/Harvard Cancer Center institutional review board following written consent.
Transcriptome and copy number analysis for LYN expression
Gene expression for LYN was determined in 57 patients with WM by validated next-generation RNA sequencing as previously reported.15 MYD88 and CXCR4 mutation status for these patients was determined as described above.13,14 Findings were compared with LYN expression in healthy donor peripheral blood B cells, memory B cells, and plasma cells. Copy number analysis for LYN was performed as before.16
HCK knockdown and overexpression studies
HCK knockdown or overexpression of WT (HCKWT) or gatekeeper mutant of HCK (HCKThr333Met) was performed using lentiviral expression vectors as previously described.4
Signaling studies
Cells were treated for 1 to 2 hours prior to PhosFlow and western blot analysis. Alexa Fluor 647-conjugated p-SYK(Tyr525/Tyr526), APC-Cy7-conjugated CD20 (BD Biosciences, San Jose, CA), and p-HCK(Tyr410) combined with DyLight 650-conjugated goat F(ab')2 anti-rabbit immunoglobulin G (IgG) antibody (Abcam, Cambridge, MA) were used for PhosFlow analysis. Western blotting was performed using antibodies to p-SYK(Tyr525/Tyr526) (R&D Systems), p-LYN(Tyr396) (GeneTex Inc), SYK, LYN, p-AKT(Ser473), AKT, p-ERK1/2(Thr202/Tyr204), ERK1/2, and HCK (Cell Signaling Technologies, MA).
Co-IP studies
Coimmunoprecipitation (co-IP) studies were performed as previously described using an HCK antibody (Cell Signaling Technologies).4
Statistical analysis
Pairwise comparisons using Wilcoxon rank sum exact test were performed for gene expression from transcriptome analysis.
Results and discussion
p-LYN expression is downregulated in MYD88 WM cells
In previous studies, the SFK LYN was shown to be a trigger for SYK activation, and in response to chronic BCR activation in ABC DLBCL.9,10 By next-generation sequencing, LYN shows variable expression in patients with WM and was significantly lower in LPCs from MYD88Mut patients who are CXCR4WT when compared with expression levels in healthy donor peripheral and memory B cells (supplemental Figure 1A). Conversely, no difference in LYN expression was observed between LPCs from MYD88Mut and CXCR4Mut patients and healthy donor peripheral and memory B cells.
To clarify the activation state of LYN in MYD88 mutated lymphoma cells, we examined p-LYN expression. MYD88Leu265Pro mutated TMD8, HBL-1, and OCI-Ly3 ABC DLBCL cells showed strong expression of p-LYN, a finding previously attributed to chronic BCR signaling.9 Conversely, p-LYN expression was low in MYD88Leu265Pro mutated BCWM.1 and MWCL-1 WM cells, and SU-DHL-2 ABC DLBCL cells that carry an MYD88Ser222Arg mutation (Figure 1A). Moreover, p-LYN expression was absent or low in CD19-selected bone marrow primary LPCs from 6 MYD88Leu265Pro patients with WM, including 3 of whom amply expressed LYN protein. Copy number analysis showed no remarkable alterations in LYN expression (supplemental Figure 1B). By comparison, LYN was uniformly expressed in CD19-selected peripheral blood B cells from 6 healthy donors and showed robust expression of p-LYN (Figure 1B). Taken together, the above findings show variable expression for LYN in patients with MYD88 mutated WM, although CXCR4 mutation status impacted LYN expression levels. Although copy number loss of LYN is common in WM, such loss is typically subclonal and not impacted by CXCR4 mutation status.16 Our copy number findings for LYN are consistent with these prior observations. Expression differences in LYN may therefore be related to epigenomic (methylation) changes that accompany CXCR4 mutation status and tumor differentiation.17,18 Indeed, lower expression levels of LYN were observed in healthy donor plasma cells (supplemental Figure 1A) consistent with the known downregulation of BCR pathway that accompanies B cell to plasma cell differentiation. As MYD88MUTCXCR4WT vs MYD88MUTCXCR4MUT WM cells show more advanced plasmacytic differentiation, the finding of lower LYN expression in this subgroup is not surprising.15,18 Importantly, the lack of SFK LYN activity observed in primary MYD88MUT WM samples (including one that was CXCR4MUT) suggests that LYN is unlikely to mediate SYK activation in MYD88Leu265Pro WM but may be its driver in MYD88Leu265Pro DLBCL cells owing to chronic active BCR signaling.9
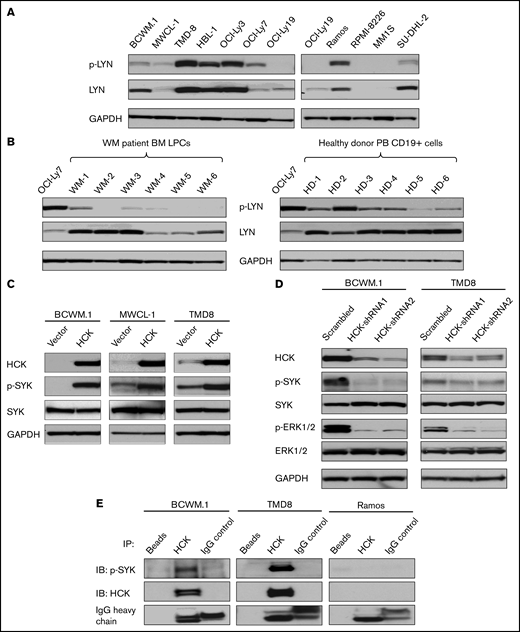
The SFK HCK activates SYK in MYD88 mutated lymphomas cells. (A) Expression of p-LYNTyr396 levels by western blot analysis in MYD88L265P BCWM.1, MWCL-1 WM cells, TMD8, HBL-1, OCI-Ly3; MYD88Ser222Arg SU-DHL-2 ABC DLBCL cells; and MYD88WT OCI-Ly7, OCI-Ly19 GCB DLBCL cells, Ramos Burkitt lymphoma cells, RPMI-8226, and MM.1S multiple myeloma cells. The expression levels of total LYN in these cells as well as protein loading control glyceraldehyde-3-phosphate dehydrogenase (GAPDH) are also shown. (B) p-LYNTyr396 levels by western blot analysis in CD19-selected bone marrow LPC from 6 MYD88Leu265Pro patients with WM of whom WM1 was also CXCR4Mut and WM2-6 was CXCR4WT, and CD19-selected peripheral blood (PB) B cells from 6 healthy donors; lysates from OCI-Ly7 GCB DLBCL cells were used for p-LYN and protein loading control. The expression of total LYN is also shown. (C) Expression of p-SYKTyr525/Tyr526 levels by western blot analysis in vector-only, HCKWT or HCKThr333Met transduced BCWM.1, MWCL-1 WM cells, and TMD8 ABC DLBCL cells. Expression levels of total HCK, SYK in these cells as well as GAPDH for protein loading control are shown. (D) Changes in p-SYKY525/526 and p-ERK1/2Thr202/Tyr204 levels following HCK knockdown with doxycycline inducible shRNA1 and shRNA2 or scrambled control vector in BCWM.1 WM and TMD8 ABC DLBCL cells. p-SYKTyr525/Tyr526 and p-ERK1/2Thr202/Tyr204 levels were detected at day 9 following 1.0 µg/mL doxycycline induction. p-ERK1/2Thr202/Tyr204, a known downstream signaling component of HCK, served as a positive control for these experiments.4 Expression levels of total HCK, SYK, and ERK1/2 as well as GAPDH for protein loading control are also shown. (E) p-SYKY525/526 protein levels by western blot analysis following a co-IP with HCK protein in MYD88Mut BCWM.1 and TMD8 cells, and MYD88WT Ramos cell lysates. Magnetic beads only and rabbit IgG were used as co-IP experimental controls. HCK total protein was also shown as an indication of the co-IP efficiency in these cells. IgG heavy chain was shown as an indication of the quantity of antibodies used in the co-IP experiments. Above experiments were performed at least twice with representative results shown. BM, bone marrow.
The SFK HCK activates SYK in MYD88 mutated lymphomas cells. (A) Expression of p-LYNTyr396 levels by western blot analysis in MYD88L265P BCWM.1, MWCL-1 WM cells, TMD8, HBL-1, OCI-Ly3; MYD88Ser222Arg SU-DHL-2 ABC DLBCL cells; and MYD88WT OCI-Ly7, OCI-Ly19 GCB DLBCL cells, Ramos Burkitt lymphoma cells, RPMI-8226, and MM.1S multiple myeloma cells. The expression levels of total LYN in these cells as well as protein loading control glyceraldehyde-3-phosphate dehydrogenase (GAPDH) are also shown. (B) p-LYNTyr396 levels by western blot analysis in CD19-selected bone marrow LPC from 6 MYD88Leu265Pro patients with WM of whom WM1 was also CXCR4Mut and WM2-6 was CXCR4WT, and CD19-selected peripheral blood (PB) B cells from 6 healthy donors; lysates from OCI-Ly7 GCB DLBCL cells were used for p-LYN and protein loading control. The expression of total LYN is also shown. (C) Expression of p-SYKTyr525/Tyr526 levels by western blot analysis in vector-only, HCKWT or HCKThr333Met transduced BCWM.1, MWCL-1 WM cells, and TMD8 ABC DLBCL cells. Expression levels of total HCK, SYK in these cells as well as GAPDH for protein loading control are shown. (D) Changes in p-SYKY525/526 and p-ERK1/2Thr202/Tyr204 levels following HCK knockdown with doxycycline inducible shRNA1 and shRNA2 or scrambled control vector in BCWM.1 WM and TMD8 ABC DLBCL cells. p-SYKTyr525/Tyr526 and p-ERK1/2Thr202/Tyr204 levels were detected at day 9 following 1.0 µg/mL doxycycline induction. p-ERK1/2Thr202/Tyr204, a known downstream signaling component of HCK, served as a positive control for these experiments.4 Expression levels of total HCK, SYK, and ERK1/2 as well as GAPDH for protein loading control are also shown. (E) p-SYKY525/526 protein levels by western blot analysis following a co-IP with HCK protein in MYD88Mut BCWM.1 and TMD8 cells, and MYD88WT Ramos cell lysates. Magnetic beads only and rabbit IgG were used as co-IP experimental controls. HCK total protein was also shown as an indication of the co-IP efficiency in these cells. IgG heavy chain was shown as an indication of the quantity of antibodies used in the co-IP experiments. Above experiments were performed at least twice with representative results shown. BM, bone marrow.
HCK modulates SYK phosphorylation in MYD88 mutated WM cells
We next investigated a direct role for the SFK HCK in mediating SYK activation. We overexpressed HCK in MYD88Leu265Pro BCWM.1 and MWCL-1 WM cell lines, and TMD8 ABC DLBCL cells. In all 3 cell lines, overexpression of HCK triggered a robust increase in phosphorylation of SYKTyr525/Tyr526 vs vector-only transduced cells (Figure 1C). Moreover, using an inducible vector system, HCK knockdown markedly reduced SYKTyr525/Tyr526 phosphorylation in BCWM.1 WM and TMD8 ABC DLBCL cells (Figure 1D). In both experiments, total SYK levels remained unchanged (Figure 1C-D).
Activated SYK is complexed with HCK in MYD88 mutated B-cell lymphoma cells
To clarify if HCK and activated SYK were present in the same signaling complex, we performed co-IP experiments using an HCK antibody in MYD88Mut BCWM.1, TMD8, and MYD88WT Ramos cells. The HCK antibody effectively pulled down p-SYK in MYD88Mut BCWM.1 and TMD8 cells, but not in MYD88WT Ramos cells (Figure 1E).
HCK kinase activity is responsible for SYK activation in MYD88 B-cell lymphoma cells
In previous studies, we showed that A419259, a potent toolbox inhibitor of HCK, shows selective killing of MYD88 mutated lymphoma cells.4 To confirm whether HCK kinase activity triggered SYK activation, we performed rescue experiments with A419259 in MYD88 mutated BCWM.1 and MWCL-1 WM and TMD8 ABC DLBCL cells expressing either HCKWT or HCK gatekeeper mutated HCKThr333Met that abrogates A419259 binding.4 BCWM.1 and MWCL-1 cells transduced to express HCKThr333Met protein showed a >2 log-fold increase in resistance to A419259 vs those transduced with either vector alone or HCKWT protein. By PhosFlow analysis, expression of HCKThr333Met but not HCKWT led to persistent activation of HCK and SYK in the presence of A419259 in BCWM.1 (Figure 2A-B) and MWCL-1 (Figure 2C-D) WM cells, and TMD8 (Figure 2E-F) ABC DLBCL cells. Consistent with these observations, treatment of primary MYD88Mut WM LPCs cells with A419259 also abrogated both HCK and SYK phosphorylation (Figure 2G).
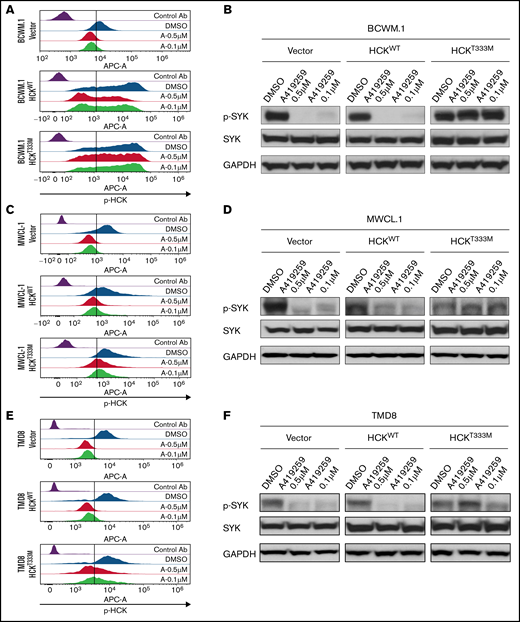
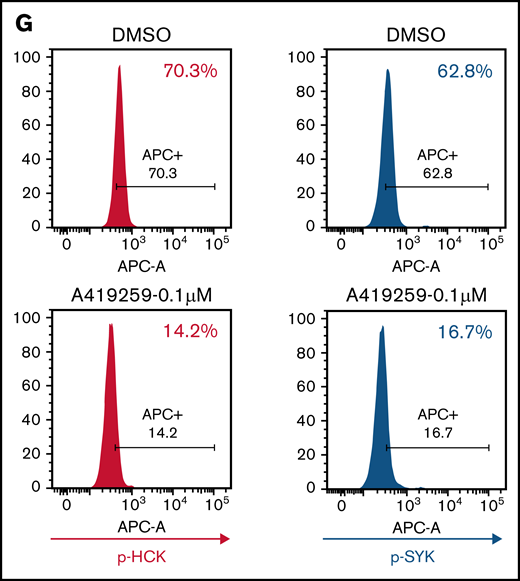
SYK activation is driven by the SFK HCK in MYD88 mutated lymphoma cells. (A) Relative p-HCKTyr410 levels by PhosFlow analysis following the treatment with dimethyl sulfoxide (DMSO) or the HCK inhibitor A419259 at the indicated concentrations for 1.0 hour in vector-only, HCKWT, or HCKThr333Met transduced BCWM.1 cells. (B) Relative p-SYKY525/526 levels by PhosFlow analysis following treatment with DMSO or A419259 at indicated concentrations for 1.0 hour in vector-only, HCKWT, or HCKThr333Met transduced BCWM.1 cells. (C) Relative p-HCKTyr410 levels by PhosFlow analysis following the treatment with DMSO or A419259 at indicated concentrations for 1.0 hour in vector-only, HCKWT, or HCKThr333Met transduced MWCL-1 cells. (D) Changes in p-SYKTyr525/Tyr526 levels following the treatment with DMSO or A419259 at indicated concentrations for 1.0 hour in vector only, HCKWT, or HCKThr333Met transduced MWCL-1 cells. The expression levels of total SYK in these cells as well as protein loading control GAPDH are also shown. (E) Relative p-HCKTyr410 levels by PhosFlow analysis following the treatment with DMSO or A419259 at indicated concentrations for 1.0 hour in vector only, HCKWT, or HCKThr333Met transduced TMD8 cells. (F) Changes in p-SYKTyr525/Tyr526 levels following the treatment with DMSO or A419259 at indicated concentrations for 1.0 hour in vector-only, HCKWT, or HCKThr333Met transduced TMD8 cells. The expression levels of total SYK in these cells as well as protein loading control GAPDH are also shown. (G) p-HCKTyr410 levels and p-SYKTyr525/Tyr526 levels by PhosFlow analysis following the treatment with DMSO or A419259 at the indicated concentrations for 1.0 hour in CD20+ gated patient with WM bone marrow lymphoplasmacytic cells. Above experiments were performed at least twice with representative results shown. Ab, antibody.
SYK activation is driven by the SFK HCK in MYD88 mutated lymphoma cells. (A) Relative p-HCKTyr410 levels by PhosFlow analysis following the treatment with dimethyl sulfoxide (DMSO) or the HCK inhibitor A419259 at the indicated concentrations for 1.0 hour in vector-only, HCKWT, or HCKThr333Met transduced BCWM.1 cells. (B) Relative p-SYKY525/526 levels by PhosFlow analysis following treatment with DMSO or A419259 at indicated concentrations for 1.0 hour in vector-only, HCKWT, or HCKThr333Met transduced BCWM.1 cells. (C) Relative p-HCKTyr410 levels by PhosFlow analysis following the treatment with DMSO or A419259 at indicated concentrations for 1.0 hour in vector-only, HCKWT, or HCKThr333Met transduced MWCL-1 cells. (D) Changes in p-SYKTyr525/Tyr526 levels following the treatment with DMSO or A419259 at indicated concentrations for 1.0 hour in vector only, HCKWT, or HCKThr333Met transduced MWCL-1 cells. The expression levels of total SYK in these cells as well as protein loading control GAPDH are also shown. (E) Relative p-HCKTyr410 levels by PhosFlow analysis following the treatment with DMSO or A419259 at indicated concentrations for 1.0 hour in vector only, HCKWT, or HCKThr333Met transduced TMD8 cells. (F) Changes in p-SYKTyr525/Tyr526 levels following the treatment with DMSO or A419259 at indicated concentrations for 1.0 hour in vector-only, HCKWT, or HCKThr333Met transduced TMD8 cells. The expression levels of total SYK in these cells as well as protein loading control GAPDH are also shown. (G) p-HCKTyr410 levels and p-SYKTyr525/Tyr526 levels by PhosFlow analysis following the treatment with DMSO or A419259 at the indicated concentrations for 1.0 hour in CD20+ gated patient with WM bone marrow lymphoplasmacytic cells. Above experiments were performed at least twice with representative results shown. Ab, antibody.
Taken together, the above studies support the activation of SYK by the SFK HCK in MYD88Mut B-cell lymphomas cells. The paucity of LYN activation in MYD88Leu2655Pro WM cells may suggest that in WM, the primary functional trigger for SYK activation, may involve the SFK HCK, whereas in MYD88Leu265Pro DLBCL both SFKs (LYN and HCK) may contribute to SYK activation. Our findings are consistent with those of Phelan et al,19 who identified a MYD88-TLR9-BCR supercomplex as a driver of BCR signaling in ABC DLBCL, and potentially extend those observations by identifying mutated MYD88 directed HCK as an enabler of SYK activation. In deference to ABC DLBCL wherein chronic active BCR signaling is known to trigger SYK through activation of LYN, we did not find evidence for LYN activation in MYD88Mut WM cells.9,10 This previously unrecognized finding may also argue against chronic active BCR signaling in WM, although further studies are needed to clarify this point. Our findings further broaden the role played by aberrant HCK expression in promoting MYD88Mut prosurvival signaling, that previously included BTK, ERK, and AKT (supplemental Figure 2). The downstream consequences of HCK may also be relevant in non-MYD88Mut driven diseases, such as mantle cell lymphoma, wherein HCK is activated.20
The recognition that HCK underlies SYK activation in MYD88Mut B-cell lymphomas may also be therapeutically relevant. A novel HCK inhibitor KIN-8194 with greater kinome selectivity and better tolerance over A419259 was more active vs ibrutinib in MYD88Mut lymphoma xenograft models.21 KIN-8194 also blocked SYK, suggesting that a broader shutdown of MYD88 prosurvival signaling may be achieved with HCK inhibitors (supplemental Figure 2). In summary, our findings show that SYK can be activated by HCK; broaden the prosurvival signaling generated by aberrant HCK expression in response to MYD88Mut; and help further establish the SFK HCK as a relevant therapeutic target in MYD88Mut B-cell lymphomas.
Acknowledgments
The authors gratefully acknowledge the generous support of Peter Bing, the International Waldenstrom’s Macroglobulinemia Foundation Legacy Grant to S.P.T., the Leukemia and Lymphoma Society grant R6507-18, the National Institutes of Health, National Cancer Institute SPORE in Multiple Myeloma grant 2P50CA100707-16A1, the Edward and Linda Nelson Fund for WM Research, the Kerry Robertson Fund for WM Research, the Bauman Family Trust, the Siegel Family Fund for WM, and the patients with WM who provided samples for these studies.
Authorship
Contribution: G.Y., M.M., and S.P.T. conceived and designed the experiments and wrote the manuscript; G.Y. and M.M. performed the data analysis; M.M. performed PhosFLow, immunoblotting, and co-IP assays; X.L. maintained cell lines and performed lentiviral expression and knockdown studies; A.K., N.T., A.G.C. and M.L.G. processed patient and healthy donor samples and performed CD19+ cell isolation; Z.R.H. provided informatics support; M.L.P. and K.V.A. provided support of BCR signaling in WM; J.W., S.J.B., and N.S.G. provided medicinal chemistry input and drug development; J.J.C., C.J.P., K.M., A.K., A.R.B., C.A.F., S.S., J.G., and S.P.T. provided patient care and obtained consent and samples; and N.C.M. and K.C.A. provided data review and input into the writing of this manuscript.
Conflict-of-interest disclosure: S.P.T. has received research funding, consulting fees, and/or honoraria from Pharmacyclics Inc, Janssen Oncology Inc, Beigene, X4 Pharmaceuticals, BMS, and Eli Lilly. J.J.C. has received research funds and/or consulting fees from Abbvie, Beigene, Janssen, Pharmacyclics, Roche, and TG Therapeutics. N.S.G. is a founder, science advisory board member, and equity holder in Syros, C4, Allorion, Jengu, B2S, Inception, EoCys, Larkspur (board member), and Soltego (board member). The Gray laboratory receives or has received research funding from Novartis, Takeda, Astellas, Taiho, Jansen, Kinogen, Arbella, Deerfield, and Sanofi. J.W. is a consultant for Soltego. S.J.B. is a member of the SAB of Adenoid Cystic Carcinoma Foundation. The Buhrlage laboratory has received research funding from AbbVie and Kinogen and in-kind resources from Novartis Institutes for Biomedical Research. A.R.B. has received consulting fees from Adaptive Biotechnologies, Beigene, CSL Behring, Karyopharm, Pharmacyclics, and Sanofi-Genzyme. C.A.F. received honoraria from Pharmacyclics and Karopharm. S.P.T., S.J.B., J.W., N.S.G., and G.Y. are named on patents owned by Dana-Farber Cancer Institute for development of HCK targeted therapeutics, including KIN-8194. G.Y. is currently employed by Blueprint. All work herein was performed before G.Y. went to Blueprint. K.C.A. is a consultant for Pfizer, Amgen, Astrazeneca, Janssen Oncology, Precision Biosciences, Mana, Window, and a Founder and stock shareholder of C4 Therapeutics, Oncopep, Raqia, and NextRNA. N.C.M. is on the advisory boards and consultant to BMS, Janssen, Amgen, Takeda, AbbVie, Oncopep, Karyopharm, Adaptive Biotechnology, Legend, Raqia, and Novartis and holds equity ownership in Oncopep. The remaining authors declare no competing financial interests.
Correspondence: Steven P. Treon, Bing Center for Waldenström's Macroglobulinemia, Dana-Farber Cancer Institute, M548, 450 Brookline Ave, Boston, MA 02115; e-mail: steven_treon@dfci.harvard.edu.

