

Key Points
Zinc is required for multimerization of mutant calreticulin, which is a requisite event for MPL binding and activation.
Zinc chelators exhibit selective toxicity against CALR-mutant MPN cell lines and primary MPN cells.

Abstract
Calreticulin (CALR) is mutated in the majority of JAK2/MPL-unmutated myeloproliferative neoplasms (MPNs). Mutant CALR (CALRdel52) exerts its effect by binding to the thrombopoietin receptor MPL to cause constitutive activation of JAK-STAT signaling. In this study, we performed an extensive mutagenesis screen of the CALR globular N-domain and revealed 2 motifs critical for CALRdel52 oncogenic activity: (1) the glycan-binding lectin motif and (2) the zinc-binding domain. Further analysis demonstrated that the zinc-binding domain was essential for formation of CALRdel52 multimers, which was a co-requisite for MPL binding. CALRdel52 variants incapable of binding zinc were unable to homomultimerize, form CALRdel52-MPL heteromeric complexes, or stimulate JAK-STAT signaling. Finally, treatment with zinc chelation disrupted CALRdel52-MPL complexes in hematopoietic cells in conjunction with preferential eradication of cells expressing CALRdel52 relative to cells expressing other MPN oncogenes. In addition, zinc chelators exhibited a therapeutic effect in preferentially impairing growth of CALRdel52-mutant erythroblasts relative to unmutated erythroblasts in primary cultures of MPN patients. Together, our data implicate zinc as an essential cofactor for CALRdel52 oncogenic activity by enabling CALRdel52 multimerization and interaction with MPL, and suggests that perturbation of intracellular zinc levels may represent a new approach to abrogate the oncogenic activity of CALRdel52 in the treatment of MPNs.
Introduction
Mutations in CALR, which encodes the endoplasmic reticulum–resident chaperone protein calreticulin (CALR) are present in the majority of JAK2/MPL-unmutated myeloproliferative neoplasms (MPNs).1,2 CALR is a calcium-binding chaperone that regulates cellular proteostasis in the endoplasmic reticulum.3 Wild-type CALR is composed of 3 domains: (1) a globular N-domain that contains lectin-, polypeptide-, and zinc-binding motifs that enable CALR chaperone activity4-6 ; (2) a P-domain that forms a hairpin structure that binds to cochaperones7,8 ; and (3) an acidic C-terminal tail involved in calcium buffering.9 MPN-associated CALR mutations are insertion-deletion mutations that result in a +1-bp frameshift that generates a neomorphic C-terminal tail encoded in an alternative reading frame, with the most common mutation being a 52-bp deletion.1,2 The resulting mutant CALR protein (CALRdel52) contains a mutant-specific C terminus in which most of the negatively charged amino acids of the wild-type protein is replaced with positively charged amino acids, such as lysine and arginine.
Multiple studies have now provided direct links between CALRdel52 and thrombopoietin receptor (MPL) signaling. Expression of CALRdel52 confers cytokine-independent growth in conjunction with increased JAK-STAT signaling in Ba/F3 and UT7 cell lines coexpressing MPL, but not erythropoietin (EPO) receptor or granulocyte-macrophage colony-stimulating factor receptor.10-12 Consistent with an essentiality of MPL in mediating CALRdel52-associated disease, CALRdel52 was unable to induce MPN-like disease in mice lacking Mpl.13 Mechanistic studies revealed that CALRdel52 physically associates with the extracellular domain of MPL10-12 through its lectin motif10,14 to activate JAK-STAT signaling and engender cytokine-independent proliferation. Moreover, CALRdel52 has also been shown to homomultimerize, and that this homomultimerization is a prerequisite for MPL binding and activation.15
In this report, we undertook a comprehensive mutagenesis screen and uncovered a new role for 3 zinc-binding histidine residues in the CALRdel52N-domain essential for CALRdel52 multimerization and MPL binding and activation. Furthermore, we found that the CALRdel52-MPL signaling complex is dependent on intracellular zinc. We show that zinc chelators were able to disrupt CALRdel52-MPL signaling complexes and attenuate JAK-STAT signaling specifically in CALR-mutant cells, and exhibited selective toxicity against CALRdel52-positive burst-forming unit erythroid (BFU-E) colonies from MPN patients. Our data reveal several additional insights into the molecular mechanism of action of CALRdel52 by identifying a central role for zinc in regulating CALRdel52 multimerization and its impact on MPL signaling.
Materials and methods
Cell culture reagents and antibodies
The 293T cells were derived from the American Type Culture Collection. MPL-expressing Ba/F3 (Ba/F3-MPL) were generated by retroviral transduction with retroviruses expressing human MPL complementary DNA. The 293T cells were maintained in Dulbecco’s modified Eagle medium (DMEM; Lonza), supplemented with 10% fetal calf serum (Sigma) and 1% penicillin-streptomycin-glutamine mix (Sigma), and cultured in 37°C with 5% CO2. Ba/F3-MPL cells were maintained in RPMI (Lonza) supplemented with 10% fetal calf serum, 1% penicillin-streptomycin-glutamine mix, and 50 ng/mL recombinant human thrombopoietin (PeproTech) in a vented flask, and cultured in 37°C with 5% CO2. For N,N,N′,N′-tetrakis(2-pyridinylmethyl)-1,2-ethanediamine (TPEN) treatment, TPEN (Sigma) was resuspended in 99% ethanol at a concentration of 20 μM and added to media at indicated concentrations for 1 to 4 hours. For clioquinol treatment, clioquinol (Sigma) was resuspended in dimethyl sulfoxide at a concentration of 40 μM and added to media at indicated concentrations for 4 hours to assess effect intracellular signaling and 48 hours to assess cell viability. For immunoblotting, primary antibodies were obtained from the following sources: mouse anti-FLAG-M2 (Sigma), rabbit anti-FLAG rabbit (Sigma), rabbit-anti-V5 (Cell Signaling), rabbit anti-CALR (clone D3E6) (Cell Signaling), rabbit anti-pSTAT5-Y694 (clone D47E7) (Cell Signaling), rabbit anti-pSTAT3-Y705 (clone D3A7) (Cell Signaling), rabbit anti-total STAT5 (clone D2O6Y) (Cell signaling), rabbit anti-STAT3 (clone124H6) (Cell Signaling), mouse anti-ACTIN (clone AC35) (Sigma), and mouse anti-MPL (clone 1.78.1) (BD Bioscience).
Alanine mutagenesis screen
Mutagenesis of iV2-LeGO-iV2-CALRdel52 plasmid DNA was performed using Q5 Hot Start High-Fidelity Master mix (New England Biolabs) according to the manufacturer’s protocols. The following thermocycling setup was used: initial denaturation 98°C for 30 seconds, 25 cycles at 98°C for 10 seconds, 50°C to 72°C for 20 seconds, and 72°C for 4 minutes, followed by a final extension at 72°C for 10 minutes. Kinase, ligase, and DpnI treatment was immediately performed following the linear amplification for 1 hour at 37°C. The kinase, ligase, and DpnI–treated reaction was used to transform competent Escherichia coli. DNA was isolated from individual colonies, and presence of alanine mutant was confirmed by Sanger sequencing. To perform functional assays for each CALR alanine variant, lentiviral supernatants were prepared for each alanine variant by cotransfection of the mutagenized LeGO-iV2-CALR variant with packaging plasmids psPAX2 and pVSV.G. After 24 hours of transfection, CALR-expressing viral supernatants were collected, filtered, and used to infect Ba/F3-MPL cells by spin infection at 2000 rpm at 37°C for 2 hours. Cells were allowed to recover by incubation with RPMI supplemented with 50 ng/mL recombinant human thrombopoietin (PeproTech) for 2 days, before cytokine withdrawal. For cytokine withdrawal, exponentially growing Ba/F3 MPL cells transduced with lentiviruses expressing various CALR variants were washed 3 times with phosphate-buffered saline (PBS) and seeded in triplicate at an initial density of 2 × 105 cells/mL in RPMI-1640 medium (Lonza) supplemented with 10% fetal bovine serum (Sigma) and 1% penicillin/streptomycin/l-glutamine (Sigma) in the absence of cytokine. Living cells were counted using hemocytometers by trypan blue exclusion assay, 3-(4,5-dimethylthiazol-2-yl)-2,5-dimethyltetrazolium bromide assays or by cell counting on a flow cytometer. The ability of each alanine variant to confer cytokine-independent growth was depicted relative to that of an unmutated LeGO-iV2-CALRdel52 control.
FLAG pulldown assays
To assess protein complex formation of heterologously expressed proteins, coimmunoprecipitations were performed in 293T cells. Cells were seeded at a density of 2 × 105 cells/mL in a 6-well plate. For assessing binary CALRdel52-MPL complex formation, DNA-cationic lipid mixtures were created by combining 1.25 μg of pCMV-SPORT6-CALR variant-expressing plasmid, 1.25 μg of pLeGO-iT2-hMPL, and 7.5 μL TransIT-LT1 transfection reagent (Mirus) in 250 μL of media-free DMEM. For assessing ternary CALRdel52-MPL complexes with 2 different tagged versions of CALRdel52, 0.625 μg of each pCMV-SPORT6-CALR variant-expressing plasmid was used and mixed with 1.25 μg of pLeGO-iT2-hMPL and 7.5 μL TransIT-LT1 transfection reagent in 250 μL media-free DMEM, as discussed previously. Mixtures were allowed to sit at room temperature for 15 minutes, and then added dropwise to 293T cells. After 24 hours, transfected 293T cells were harvested, washed with PBS twice, and then cells were collected and resuspended in 450 μL NP40 lysis buffer and incubated on ice for 30 minutes. Lysates were cleared by centrifugation at 17000g for 10 minutes at 4°C, and 10% (usually ∼50 μL) was set aside as the total lysate “input” fraction and the remainder used for pulldown assays. Protein G bead-antibody mixtures were generated by initially washing 50 μL of magnetic Protein G Surebeads (BioRad Laboratories) per sample with PBS with 0.1% Tween-20 [PBST]) 3 times using a magnet. One microliter of FLAG M2 antibody (Sigma) was diluted with PBST and added to the beads and incubated for 15 minutes with inversion at room temperature. FLAG M2-Protein G beads were washed with PBST and incubated with lysate supernatants for 1 hour at room temperature and mixed by inversion. Lysate-bead mixtures were washed 3 times by magnetic sorting, and beads were resuspended in 100 μL of 1X SDS Laemmli buffer and eluted by incubating the beads for 10 minutes at 70°C. Both pulldown fractions and lysates were analyzed by Western immunoblotting, as described in supplemental Information.
Intracellular phosphoprotein flow cytometry analysis
Ba/F3-MPL cells expressing CALR variants were cultured in RPMI supplemented with 10% fetal bovine serum, 5% penicillin/streptomycin, and 5% l-Glutamine for 24 hours in the absence of cytokines. Harvested cells were transferred into flow tubes and washed with PBS + 1% bovine serum albumin (BSA) twice to remove residual RPMI media. These cells were fixed and permeabilized using Fix&Perm Kit (Nordic MUbio). Staining was carried out in PBS 1% BSA for 1 hour at room temperature with PE Mouse anti-Stat3 (pY705) and Alexa Fluor 647 anti-Stat5 (pY694) (BD Biosciences). Cells were washed with PBS with 1% BSA and samples were analyzed using a CytoFLEX Flow Cytometer (Beckman Coulter Life Sciences). Data analysis was performed with CytExpert V2.2.
Confocal microscopy and FRET analysis
The 293T cells were seeded into 35mm, high, μ-Dish with an ibidi polymer coverslip bottom and were incubated overnight at 37°C. Transient co-transfection of pSport6-Cherry-CALR variant-FLAG with MSCV-GFP-MPL in a 1:1 ratio was performed using TransIT-LT1 (Mirus). Following 24 hours post-transfection, cells were gently washed and resuspended with phenol red-free DMEM media, and cells were imaged using Zeiss LSM 880 inverted confocal microscopy. Fluorescence resonance energy transfer (FRET) was measured by exciting sample at donor excitation of 488 nm and measuring the acceptor excitation absorbance at 610 nm. FRET intensity was calculated by using sensitized emission.
TPEN treatment of clonal erythroblasts from MPN patients
MPN patients were diagnosed according to the British Committee for Standards in Hematology guidelines.16 UK patient samples were obtained under local ethics approval (Cambridge and Eastern Region Ethics Committee) or as part of the UK Medical Research Council PT-1 trial.17 Written informed consent was obtained from all patients in accordance with the Declaration of Helsinki. For local Cambridge patients, samples were taken at the time of the initial patient visit on referral to the specialist MPN clinic at the Addenbrooke’s Hospital in Cambridge, United Kingdom. Venous blood samples (20 mL) were collected from each patient and peripheral blood mononuclear cells (PBMCs) were isolated using Lymphoprep (Axis Shield PLC) according to the manufacturer’s protocols and plated in complete MethoCult medium (H4435; STEMCELL Technologies) at a density of 3 × 105 cells/mL and incubated at 37°C and 5% CO2 for 14 days. When indicated, MethoCult was supplemented with 5 μM TPEN (Sigma). After 14 days, colonies were plucked into 20 μL PBS. For each colony, 5 μL was denatured at 95°C for 15 minutes to release cellular DNA and subjected to polymerase chain reaction using primers to amplify exon 9 of the CALR gene (forward primer: 5′-GCA GCA GAG AAA CAA ATG AAG G-3′; reverse primer: 5′-AGA GTG GAG GAG GGG AAC AA-3′). Polymerase chain reaction conditions were an initial preheating at 94°C for 10 minutes was followed by denaturation at 94°C for 30 seconds, annealing at 64°C for 30 seconds, and extension at 72°C for 30 seconds for 40 cycles, followed by a final extension at 72°C for 7 minutes. Amplicons were submitted for Sanger sequencing to identify colony genotypes.
Results
Alanine mutagenesis screen of CALRdel52N-domain demonstrates residues associated with lectin and zinc-binding activity are essential for oncogenic activity
We and others previously reported the lectin motif within the CALRdel52N-domain was essential for MPL binding.10,14 To resolve other regions within the N-domain potentially necessary for oncogenic activity, we undertook an alanine mutagenesis screen of the entire N-domain by screening for the ability of single-residue loss-of-function variants to engender cytokine-independence in MPL-expressing Ba/F3 (Ba/F3-MPL) cells. We observed that the majority of the 177 single-residue alanine mutants did not affect CALRdel52 activity, with only 9 residues abrogating cytokine independence by a factor greater than 1 standard deviation from the mean when mutated (Figure 1A). Specifically, CALRdel52 harboring C105A, K111A, G133A, or D135A mutations exhibited total abrogation of cytokine independence (Figure 1B, left), decreased STAT3/5 phosphorylation as assessed by intracellular flow cytometry (Figure 1C) and immunoblotting (Figure 1D), and an inability to bind MPL in FLAG-pulldown assays (Figure 1E). These residues have previously been implicated in the lectin activity of wild-type CALR,18 and D135 specifically has previously been shown by us and others to be required for CALRdel52 oncogenic activity.10,14 In addition, CALRdel52 harboring Y109A and Y128A mutations only exhibited partial loss of cytokine independence (Figure 1B, right) and exhibited no effect on STAT3/5 phosphorylation or MPL binding (Figure 1C-E), despite these residues also having been implicated in wild-type CALR lectin activity. Of note, other residues thought to be essential for CALR lectin motif such as M131 and I147 did not exhibit any effect in our screen (Figure 1A). Overall, these data accord with previous findings that the lectin motif of CALRdel52 is essential for oncogenic activity, but suggest that different residues within the lectin motif of CALRdel52 may have differential effects in facilitating its oncogenic activity.
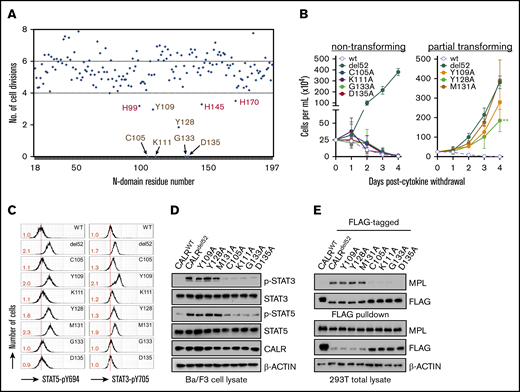
The lectin-dependent function of CALRdel52is required for cytokine-independent growth. (A) Alanine mutagenesis screen to assess ability of 177 CALRdel52 single-residue mutants to confer cytokine-independent growth of Ba/F3-MPL cells, as depicted on y-axis by number of cell divisions following 3 days. The mutagenized residue is represented on the x-axis. The 9 residues that are >1 standard deviation away from mean are indicated with arrows. Putative lectin residues are denoted in brown; putative zinc-binding residues are denoted in red. (B) Growth curves of Ba/F3-MPL cells expressing wild-type CALR, CALRdel52, or lectin motif variants demonstrate total (left) or partial (right) impairment of cytokine-independent growth. Testing for statistical significance was performed using a Student t test (*P < .05; **P < .01; ***P < .001). (C) Intracellular phosphorylation flow cytometry and (D) Western immunoblotting demonstrate diminished Stat3 and Stat5 phosphorylation in Ba/F3-MPL cells expressing nontransforming CALRdel52 lectin variants. Numbers in red indicate ratio of mean fluorescence intensity for each sample relative to isotype control. The data are representative of 2 independent experiments. (E) Immunoblotting of FLAG-immunoprecipitated proteins from 293T cells cotransfected with FLAG-tagged CALRdel52 lectin variants and MPL-expressing vector demonstrates impaired MPL binding capacity by nontransforming lectin variants.
The lectin-dependent function of CALRdel52is required for cytokine-independent growth. (A) Alanine mutagenesis screen to assess ability of 177 CALRdel52 single-residue mutants to confer cytokine-independent growth of Ba/F3-MPL cells, as depicted on y-axis by number of cell divisions following 3 days. The mutagenized residue is represented on the x-axis. The 9 residues that are >1 standard deviation away from mean are indicated with arrows. Putative lectin residues are denoted in brown; putative zinc-binding residues are denoted in red. (B) Growth curves of Ba/F3-MPL cells expressing wild-type CALR, CALRdel52, or lectin motif variants demonstrate total (left) or partial (right) impairment of cytokine-independent growth. Testing for statistical significance was performed using a Student t test (*P < .05; **P < .01; ***P < .001). (C) Intracellular phosphorylation flow cytometry and (D) Western immunoblotting demonstrate diminished Stat3 and Stat5 phosphorylation in Ba/F3-MPL cells expressing nontransforming CALRdel52 lectin variants. Numbers in red indicate ratio of mean fluorescence intensity for each sample relative to isotype control. The data are representative of 2 independent experiments. (E) Immunoblotting of FLAG-immunoprecipitated proteins from 293T cells cotransfected with FLAG-tagged CALRdel52 lectin variants and MPL-expressing vector demonstrates impaired MPL binding capacity by nontransforming lectin variants.
Next, we examined the other residues identified in our screen that have not previously been associated with glycan binding. We observed that CALRdel52 harboring H99A, H145A, and H170A mutations (1His) exhibited partial abrogation of cytokine independence (Figure 1A and Figure 2A, left). Because these residues have been reported to need to function in a concerted manner to coordinate zinc,19 we tested whether combined loss of multiple residues would have more deleterious consequences on CALRdel52 activity. Indeed, we observed that loss of any 2 or all 3 histidine residues in combination (2His and 3His, respectively) led to significant impairment of cytokine-independent growth (Figure 2A, middle and right), in conjunction with decreased pSTAT3/5 phosphorylation (Figure 2B-C).
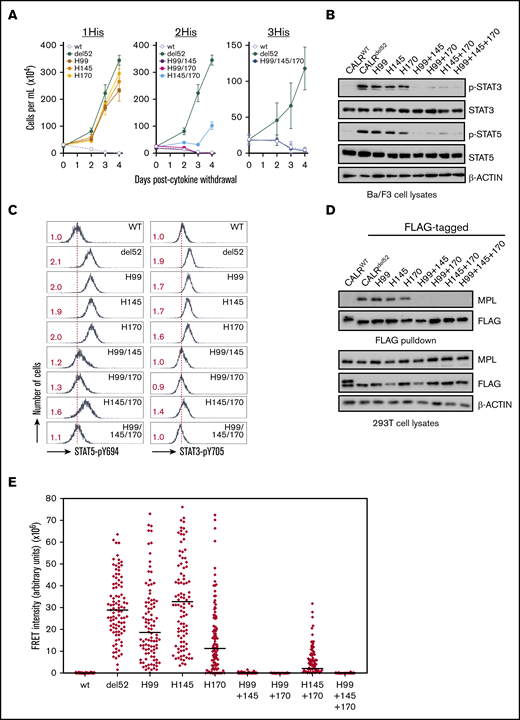
A triad of zinc-binding histidine residues in CALRdel52are required for cytokine-independent growth. (A) Growth curves in Ba/F3-MPL cells expressing histidine-deficient CALRdel52 variants harboring loss of either 1 histidine (1His), 2 histidines (2His), or 3 histidines (3His). Immunoblotting (B) and intracellular phospho-flow cytometry (C) demonstrate diminished Stat3 and Stat5 phosphorylation in of Ba/F3-MPL cells expressing 2His- and 3His-CALRdel52 variants. Numbers in red indicate ratio of mean fluorescence intensity for each sample relative to isotype control. The data are representative of 2 independent experiments. (D) Immunoblotting of FLAG-immunoprecipitated proteins from 293T cells cotransfected with histidine-deficient FLAG- CALRdel52 variants and MPL-expressing vectors demonstrates abolished MPL binding capacity by 2His- and 3His-CALRdel52 variants. (E) Single-cell data for FRET intensity. FRET fluorescence units depict energy transfer between mCherry-tagged histidine-deficient CALRdel52 protein and MPL-GFP in arbitrary fluorescence units for a single cell. For each experimental condition, 100 cells were analyzed.
A triad of zinc-binding histidine residues in CALRdel52are required for cytokine-independent growth. (A) Growth curves in Ba/F3-MPL cells expressing histidine-deficient CALRdel52 variants harboring loss of either 1 histidine (1His), 2 histidines (2His), or 3 histidines (3His). Immunoblotting (B) and intracellular phospho-flow cytometry (C) demonstrate diminished Stat3 and Stat5 phosphorylation in of Ba/F3-MPL cells expressing 2His- and 3His-CALRdel52 variants. Numbers in red indicate ratio of mean fluorescence intensity for each sample relative to isotype control. The data are representative of 2 independent experiments. (D) Immunoblotting of FLAG-immunoprecipitated proteins from 293T cells cotransfected with histidine-deficient FLAG- CALRdel52 variants and MPL-expressing vectors demonstrates abolished MPL binding capacity by 2His- and 3His-CALRdel52 variants. (E) Single-cell data for FRET intensity. FRET fluorescence units depict energy transfer between mCherry-tagged histidine-deficient CALRdel52 protein and MPL-GFP in arbitrary fluorescence units for a single cell. For each experimental condition, 100 cells were analyzed.
To ensure that these results are specific to these individual zinc-binding histidine residues and not from nonspecific effect on CALRdel52 stability, we also tested the effects of CALRdel52 in which H42, which is predicted to lie on the opposite face of CALRdel52 and is not thought to function in concert with H99, H145, or H170 to bind zinc,18 is mutated. We observed that CALRdel52 variants harboring H42A mutations or of H42A in combination with H99, H145, or H170 were unaffected in their ability to confer cytokine-independent growth (supplemental Figure 1A) and retained the capacity to promote STAT3/5 phosphorylation (supplemental Figure 1B-C).
Finally, we also tested whether histidine-deficient CALRdel52 variants could physically associate with MPL by co-immunoprecipitation assays. We observed that 1His-CALRdel52 variants retained the ability to bind to MPL in FLAG pulldowns, but not 2His or 3His variants (Figure 2D). We also tested the ability of histidine-deficient CALRdel52 variants to colocalize intracellularly with MPL using FRET. To achieve this, histidine-deficient CALRdel52 variants were generated fused to an mCherry fluorescent marker and introduced to 293T cells in conjunction with an MPL-GFP fusion protein; colocalization was measured by quantification of energy transfer of the mCherry donor to the GFP acceptor. Significant FRET was observed for mCherry-tagged CALRdel52 and MPL-GFP indicative of intracellular colocalization, whereas minimal FRET was observed for mCherry-tagged wild-type CALR and MPL-GFP (supplemental Figure 2). Analysis of 100 cells for each histidine-deficient CALRdel52 variants indicated that MPL colocalized strongly with 1His-CALRdel52 variants, but not with 2His and 3His variants (Figure 2E).
Cumulatively, functional analysis of single-residue CALRdel52N-domain variants were able to identify 2 classes of residues that are important for mediating the ability of CALRdel52 to bind and activate MPL and confer cytokine independence: (1) residues associated with lectin motif and (2) histidine residues putatively associated with zinc binding.
Histidine-mutated CALRdel52 is incapable of homomultimerization
A recent study detected CALRdel52 homomultimerization by pulldown of distinct CALRdel52 molecules tagged with 2 different epitopes.15 We used the same strategy to test the multimerization capacity of lectin-deficient or histidine-mutated CALRdel52. We confirmed these findings by assessing the ability of FLAG-tagged CALRdel52 (bait) to complex and co-immunoprecipitate with a V5-tagged CALRdel52 species (prey) in 293T cells (Figure 3A), and demonstrated that CALRdel52 is capable of interacting with CALRdel52 but exhibited impaired ability to bind to wild-type CALR (Figure 3B). Using this experimental setup, we next tested the multimerization capacity of lectin-deficient or histidine-mutated CALRdel52. We observed that the lectin-deficient CALRdel52 and all 3 1His-CALRdel52 variants retained multimerization capacity equivalent to that of CALRdel52 (Figure 3B-C). In contrast, we observed that the 2His- and 3His-CALRdel52 were impaired in their capacity to multimerize with CALRdel52 (Figure 3C). To ensure that these activities were not an artifact of assessing binding with unmutated CALRdel52, we also tested the ability of lectin-deficient or histidine-mutated CALRdel52 to multimerize with another lectin-deficient or histidine-mutated CALRdel52, respectively, and form homomultimers. Consistent with our earlier data, we observed that the lectin-deficient CALRdel52-D135A retained the ability to homomultimerize with another CALRdel52-D135A, but the histidine-mutated 3His-CALRdel52 was not able to homomultimerize with another 3His-CALRdel52 (Figure 3D). Our data therefore accord with previous reports that CALRdel52 forms homomultimers, but our data show that homomultimerization is necessary but not sufficient for MPL binding.
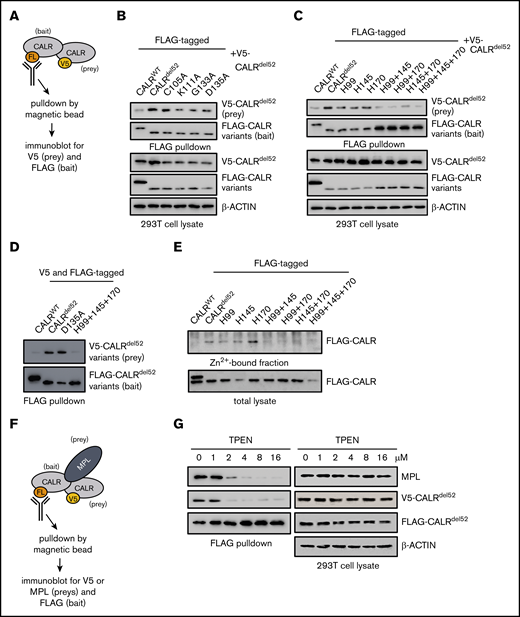
Zinc binding by CALRdel52is required for homomultimerization and enables MPL activation. (A) Schematic depicting co-immunoprecipitation assay for detecting FLAG-tagged and V5-tagged CALRdel52 multimers. (B) FLAG-pulldown assays demonstrating CALRdel52 lectin variants retain ability to bind to V5-CALRdel52. (C) FLAG-pulldown assays demonstrating 2His- and 3His-CALRdel52 are compromised in ability to bind V5-CALRdel52 compared with 1His-CALRdel52. (D) FLAG-pulldown assays of cells transfected with FLAG- and V5-tagged CALRWT (lane 1), CALRdel52 (lane 2), CALRdel52 D135A lectin variant (lane 3), or 3His-CALRdel52 variant (lane 4) demonstrating that CALRdel52 and CALRdel52 D135A are capable of homomultimerization, but not CALRWT and 3His-CALRdel52. (E) Affinity chromatography demonstrating 2His- and 3His-CALRdel52 bind less efficiently to zinc resin compared with 1His-CALRdel52. (F) Schematic depicting co-immunoprecipitation assay for detecting heteromeric complexes comprising FLAG-tagged and V5-tagged CALRdel52 proteins and MPL. (G) FLAG-pulldown assays demonstrating zinc chelator TPEN treatment disrupts CALRdel52 multimerization and MPL binding in 293T cells.
Zinc binding by CALRdel52is required for homomultimerization and enables MPL activation. (A) Schematic depicting co-immunoprecipitation assay for detecting FLAG-tagged and V5-tagged CALRdel52 multimers. (B) FLAG-pulldown assays demonstrating CALRdel52 lectin variants retain ability to bind to V5-CALRdel52. (C) FLAG-pulldown assays demonstrating 2His- and 3His-CALRdel52 are compromised in ability to bind V5-CALRdel52 compared with 1His-CALRdel52. (D) FLAG-pulldown assays of cells transfected with FLAG- and V5-tagged CALRWT (lane 1), CALRdel52 (lane 2), CALRdel52 D135A lectin variant (lane 3), or 3His-CALRdel52 variant (lane 4) demonstrating that CALRdel52 and CALRdel52 D135A are capable of homomultimerization, but not CALRWT and 3His-CALRdel52. (E) Affinity chromatography demonstrating 2His- and 3His-CALRdel52 bind less efficiently to zinc resin compared with 1His-CALRdel52. (F) Schematic depicting co-immunoprecipitation assay for detecting heteromeric complexes comprising FLAG-tagged and V5-tagged CALRdel52 proteins and MPL. (G) FLAG-pulldown assays demonstrating zinc chelator TPEN treatment disrupts CALRdel52 multimerization and MPL binding in 293T cells.
Zinc is essential for CALRdel52-MPL complex formation and downstream signaling
H99, H145, and H170 have previously been implicated in zinc-binding capacity of wild-type CALR.19 Therefore, to confirm that loss of zinc coordination underpinned the loss of oncogenic capacity of 2His- and 3His-CALRdel52, we performed affinity chromatography using a zinc resin and found that CALRdel52 and 1His-CALRdel52 could readily be retained in the zinc-bound fraction, but that wild-type CALR, 2His-, and 3His-CALRdel52 exhibited diminished zinc binding (Figure 3E). That wild-type CALR did not bind to the zinc resin is somewhat surprising because wild-type CALR has previously been shown to bind zinc.10 This may suggest that the assay used in our study was not sufficiently sensitive to detect the wild-type CALR-zinc interaction, and that there may be differences in the avidity of wild-type and mutant CALR monomers to zinc and/or that CALRdel52 (but not wild-type CALR) forms multimers or aggregates when bound to zinc, resulting in greater recovery upon elution. Cumulatively, these data are consistent with the finding that 2His- and 3His-CALRdel52 exhibit loss of zinc binding.
These data therefore suggested that binding to zinc could be required for CALRdel52 multimerization and MPL binding. To test this, FLAG- and V5-tagged CALRdel52 and MPL were expressed in 293T cells, treated with agents that modulate zinc levels and FLAG pulldown was performed to assess the extent of CALRdel52 multimerization and MPL binding in heteromeric complexes (Figure 3F). To modulate zinc levels, TPEN was used as a membrane-permeable ion chelator with a high affinity for zinc.20 Treatment with TPEN led to decreased CALRdel52 multimerization and impaired MPL binding (Figure 3G), suggesting that zinc is required for CALRdel52 to multimerize and interact with MPL.
These data would predict that zinc chelation could abolish cytokine independence and JAK-STAT signaling specifically in CALRdel52-expressing hematopoietic cells but potentially not in cells expressing other MPN oncogenes. We therefore tested the effect of TPEN on cytokine-independent growth of CALRdel52-expressing Ba/F3-MPL cells (Ba/F3-MPL-CALRdel52), and observed that TPEN treatment led to increased cytotoxicity (Figure 4A) and attenuated STAT3/5 phosphorylation (Figure 4B; supplemental Figure 3), which were correlated with decreased interaction between CALRdel52 and MPL (Figure 4C). Moreover, the effect on JAK-STAT signaling was specific to CALRdel52 because Ba/F3 cells transformed with the alternate MPN oncogene MPLW515L (Ba/F3-MPLW515L) was less sensitive to TPEN-induced cytotoxicity and reduction of STAT3/5 phosphorylation (Figure 4A-B; supplemental Figure 3), despite equivalent reduction of intracellular zinc (supplemental Figure 4A-C). Similar results were obtained using a cytokine-dependent megakaryocytic cell line UT7 coexpressing MPL. TPEN induced CALRdel52-specific cytotoxicity in UT7 cells (supplemental Figure 5A), disassociation of MPL-CALRdel52 complexes (supplemental Figure 5B), and selective abrogation of JAK-STAT signaling in CALRdel52-expressing cells (supplemental Figure 5C).
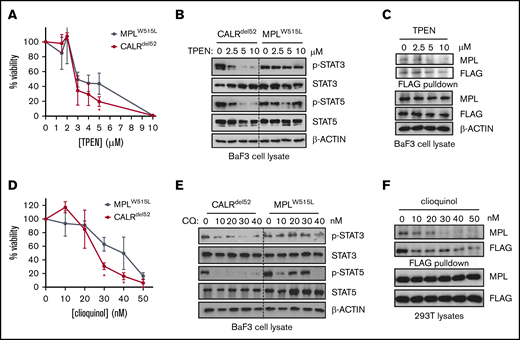
Zinc chelation abrogates CALRdel52-induced JAK-STAT signaling in hematopoietic cell lines. (A) Viability of BaF3-MPL cells expressing CALRdel52 or MPLW515L following treatment with TPEN for 48 hours. Each point represents the mean of 3 independent cultures. Testing for statistical significance was performed using a Student t test (*P < .05). (B) Immunoblotting demonstrates decreased Stat3 and Stat5 phosphorylation status following TPEN treatment of 4 hours in CALRdel52-expressing Ba/F3-MPL cells but not in Ba/F3-MPLW515L cells. (C) FLAG-pulldown assays demonstrating zinc chelator TPEN treatment disrupts CALRdel52-MPL binding in CALRdel52-expressing Ba/F3-MPL cells. (D) Viability of BaF3-MPL cells expressing CALRdel52 or MPLW515L following treatment with clioquinol (CQ) for 48 hours. Cell viability was quantified by 3-(4,5-dimethylthiazol-2-yl)-2,5-dimethyltetrazolium bromide assays. Each point represents the mean of 3 independent cultures. The data are representative of at least 2 independent experiments. Testing for statistical significance was performed using Student t test (*P < .05). (E) Immunoblotting demonstrates decreased Stat3 and Stat5 phosphorylation status following CQ treatment of 4 hours in CALRdel52-expressing Ba/F3-MPL cells but not in Ba/F3-MPLW515L cells. (F) Immunoblotting of FLAG-immunoprecipitated proteins in 293T cells expressing CALRdel52 and MPL demonstrates disruption of CALRdel52-MPL binding following CQ treatment of 4 hours.
Zinc chelation abrogates CALRdel52-induced JAK-STAT signaling in hematopoietic cell lines. (A) Viability of BaF3-MPL cells expressing CALRdel52 or MPLW515L following treatment with TPEN for 48 hours. Each point represents the mean of 3 independent cultures. Testing for statistical significance was performed using a Student t test (*P < .05). (B) Immunoblotting demonstrates decreased Stat3 and Stat5 phosphorylation status following TPEN treatment of 4 hours in CALRdel52-expressing Ba/F3-MPL cells but not in Ba/F3-MPLW515L cells. (C) FLAG-pulldown assays demonstrating zinc chelator TPEN treatment disrupts CALRdel52-MPL binding in CALRdel52-expressing Ba/F3-MPL cells. (D) Viability of BaF3-MPL cells expressing CALRdel52 or MPLW515L following treatment with clioquinol (CQ) for 48 hours. Cell viability was quantified by 3-(4,5-dimethylthiazol-2-yl)-2,5-dimethyltetrazolium bromide assays. Each point represents the mean of 3 independent cultures. The data are representative of at least 2 independent experiments. Testing for statistical significance was performed using Student t test (*P < .05). (E) Immunoblotting demonstrates decreased Stat3 and Stat5 phosphorylation status following CQ treatment of 4 hours in CALRdel52-expressing Ba/F3-MPL cells but not in Ba/F3-MPLW515L cells. (F) Immunoblotting of FLAG-immunoprecipitated proteins in 293T cells expressing CALRdel52 and MPL demonstrates disruption of CALRdel52-MPL binding following CQ treatment of 4 hours.
Next, to ensure this effect was generalizable to other zinc chelators, we also tested whether clioquinol, which has also been reported to have zinc-chelating activity,21 had a similar effect. As with TPEN, treatment of cells with clioquinol also led to preferential CALRdel52 cytotoxicity in Ba/F3-MPL cells (Figure 4D) and abrogation of JAK-STAT signaling (Figure 4E). Moreover, clioquinol could also promote disassociation of MPL-CALRdel52 complexes in 293T cells (Figure 4F) These data suggest that sensitivity to zinc homeostasis is a feature of CALRdel52-induced MPN.
TPEN preferentially eradicates CALR-mutant primary MPN cells
Finally, we tested the effect of zinc chelation on CALR-mutant primary MPN cells. We initially performed tests to identify TPEN concentrations which would affect growth of wild-type BFU-Es grown from PBMCs in semisolid medium, and observed no impaired growth of wild-type colonies at TPEN dosages lower than at 3 μM (Figure 5A). Next, PBMCs from 5 CALRdel52-mutated myelofibrosis (MF) patients (with allele burdens ranging from 20% to 73%) were grown in semisolid medium supplemented with 2.5 μM TPEN. After 14 days, BFU-Es were plucked and genotyped for the presence of CALR mutation by Sanger sequencing. In 4 of 5 MF patients examined, TPEN treatment led to decreased growth of CALRdel52-positive BFU-E colonies compared with autologous wild-type colonies (Figure 5B), with the fraction of mutant colonies in TPEN-treated cultures reduced to 74% compared with untreated cultures (P = .053) (Figure 5C). The effect of TPEN appeared to be specific to CALR-mutant cells because no selective effect was observed on JAK2V617F-mutant BFU-Es in response to TPEN in 4 JAK2-mutant MF patients examined (Figure 5D-E), and no CALRdel52 mutant-specific eradication was observed following treatment with an established MPN treatment, hydroxyurea (Figure 5F-G). Cumulatively, these data provide a proof of principle that zinc chelation selectively affects the growth of CALRdel52-positive patient-derived BFU-Es and has the potential to be harnessed as a therapeutic modality in MPN patients.
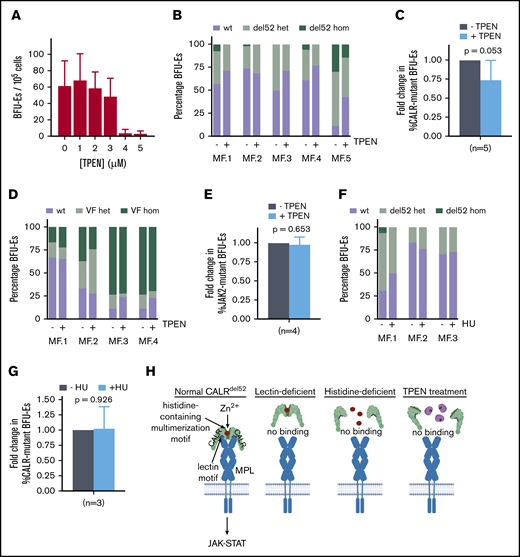
Zinc chelation exhibits selective cytotoxicity against CALR-mutant cells from MPN patients. (A) BFU-E colony counts of PBMCs from healthy volunteers grown in methylcellulose in the presence of 0 to 5 μM TPEN. (B) PBMCs from 5 myelofibrosis patients (allele burden ranging from 20% to 73%) were cultured in the presence and absence of 2.5 μM TPEN, and BFU-Es genotyped for the presence of CALR del52 mutation after 14 days. Proportion of colonies with wild-type CALR (wt), heterozygous for CALR del52 mutation (del52 het), or homozygous for CALR del52 mutation (del52 hom) cultured in the presence and absence of 2.5 μM TPEN. At least 20 colonies were genotyped for each patient. (C) Average fold change of CALR-mutant colonies (including heterozygous and homozygous colonies) for 5 MF patients examined following TPEN treatment relative to untreated cultures, which was arbitrarily designated as 1. (D) Proportion of colonies with JAK2 (wt), heterozygous for JAK2V617F mutation (VF het), or homozygous for JAK2V617F mutation (VF hom) cultured in the presence and absence of 2.5 μM TPEN. At least 20 colonies were genotyped for each patient. (E) Average fold change of JAK2-mutant colonies (including heterozygous and homozygous colonies) for 4 MF patients examined following TPEN treatment relative to untreated cultures, which was arbitrarily designated as 1. (F) Proportion of colonies with CALR (wt), heterozygous for CALR del52 mutation (del52 het), or homozygous for CALR del52 mutation (del52 hom) cultured in the presence and absence of 2 μM hydroxyurea (HU). At least 15 colonies were genotyped for each patient. (G) Average fold change of CALR-mutant colonies (including heterozygous and homozygous colonies) for 3 MF patients examined following HU treatment relative to untreated cultures, which was arbitrarily designated as 1. (H) Model depicting interplay between zinc, CALRdel52 multimerization and MPL activation. (C-G) Testing for statistical significance was performed using Student t test (*P < .05).
Zinc chelation exhibits selective cytotoxicity against CALR-mutant cells from MPN patients. (A) BFU-E colony counts of PBMCs from healthy volunteers grown in methylcellulose in the presence of 0 to 5 μM TPEN. (B) PBMCs from 5 myelofibrosis patients (allele burden ranging from 20% to 73%) were cultured in the presence and absence of 2.5 μM TPEN, and BFU-Es genotyped for the presence of CALR del52 mutation after 14 days. Proportion of colonies with wild-type CALR (wt), heterozygous for CALR del52 mutation (del52 het), or homozygous for CALR del52 mutation (del52 hom) cultured in the presence and absence of 2.5 μM TPEN. At least 20 colonies were genotyped for each patient. (C) Average fold change of CALR-mutant colonies (including heterozygous and homozygous colonies) for 5 MF patients examined following TPEN treatment relative to untreated cultures, which was arbitrarily designated as 1. (D) Proportion of colonies with JAK2 (wt), heterozygous for JAK2V617F mutation (VF het), or homozygous for JAK2V617F mutation (VF hom) cultured in the presence and absence of 2.5 μM TPEN. At least 20 colonies were genotyped for each patient. (E) Average fold change of JAK2-mutant colonies (including heterozygous and homozygous colonies) for 4 MF patients examined following TPEN treatment relative to untreated cultures, which was arbitrarily designated as 1. (F) Proportion of colonies with CALR (wt), heterozygous for CALR del52 mutation (del52 het), or homozygous for CALR del52 mutation (del52 hom) cultured in the presence and absence of 2 μM hydroxyurea (HU). At least 15 colonies were genotyped for each patient. (G) Average fold change of CALR-mutant colonies (including heterozygous and homozygous colonies) for 3 MF patients examined following HU treatment relative to untreated cultures, which was arbitrarily designated as 1. (H) Model depicting interplay between zinc, CALRdel52 multimerization and MPL activation. (C-G) Testing for statistical significance was performed using Student t test (*P < .05).
Discussion
Multiple studies have now provided evidence that CALR mutations coopt the MPL-JAK2 signaling axis to engender an MPN phenotype and that this is achieved by direct interaction between CALRdel52 and the MPL extracellular domain.10-12 In this report, we attempt to provide further molecular insights into how the CALRdel52-MPL binding event is governed. We identified a triad of zinc-binding histidine residues within the CALRdel52N-domain, which is required for both CALRdel52 homomultimerization and MPL binding, and for engendering the MPN phenotype. To our knowledge, our report is the first demonstrating of the importance of zinc as an essential cofactor in mediating MPN oncogenic signaling.
The precise mechanism by which zinc enables CALRdel52 multimerization and MPL activation remains unclear. It was somewhat surprising that 2His- and 3His-CALRdel52 molecules were incapable of binding to MPL despite the presence of an intact lectin motif. We can envision 2 potential explanations for this finding. One possibility is that zinc binding is required to ensure proper conformation of the lectin motif, and that the absence of zinc renders the lectin motif nonfunctional. Our data point toward a second possibility, whereby CALRdel52 cannot bind to MPL as a monomer but rather needs to engage MPL as a multimer, and that zinc is necessary to mediate this multimerization. Under this scenario, binding by CALRdel52 to MPL could require both an intact lectin motif and the capacity for zinc-dependent homomultimerization (Figure 5H). Indeed, there is precedent for a role for ligand multimerization in cytokine signaling. For example, EPO has been found to form high-molecular-weight species when purified22 and EPO dimers have been shown to exhibit more than 26-fold higher biological activity than its monomeric form.23 Finally, it is possible that these 2 possibilities are not mutually exclusive because mutant CALR multimerization could affect the activity of the lectin motif through an unknown mechanism to enhance its ability to bind MPL. Atomic level resolution of the structure CALRdel52-MPL complex will likely be required to resolve these outstanding issues.
Finally, our findings have potential therapeutic implications. Our data show that chemical chelation of intracellular zinc by 2 different heavy metal chelators was able to promote disassembly of the CALRdel52-MPL complex and cause attenuation of JAK-STAT signaling in CALR-mutant cells. Moreover, zinc chelation led to growth inhibition of CALRdel52-positive erythroblasts from primary MF samples. These data are, to our knowledge, the first to implicate heavy metal homeostasis in MPN pathogenesis and have the potential to support the development or repurposing of zinc or heavy metal chelators for treatment of CALR-mutated MPNs. Moreover, these data also open up the possibility that agents that interfere with CALRdel52 multimerization could also be an effective therapeutic strategy for impairing CALRdel52 oncogenic activity.
In conclusion, our data provide several novel insights into the molecular mechanism by which CALRdel52 interacts with MPL to induce MPN. Specifically, we (1) uncover a triad of zinc-binding histidines within CALRdel52 required for its oncogenic activity; (2) provide evidence that zinc-dependent homomultimerization of CALRdel52 underlies its ability to bind MPL; and (3) determine that reduction of intracellular zinc abrogates the oncogenic activity of CALRdel52 and could represent a new therapeutic modality in the treatment of CALR-mutant MPNs.
Data and reagents will be made available to other investigators upon request to the corresponding author at e.chen@westminster.ac.uk.
Acknowledgments
This work was supported by a Leuka John Goldman fellowship (JGF 2016/001) (E.C.), an Academy of Medical Science Springboard Award (SBF001\1006) (E.C.), a grant from the National Institutes of Health, National Heart, Lung, and Blood Institute (R01HL131835) (A.M.), MPN Research Foundation (A.M.), Gabrielle’s Angel Foundation for Cancer Research (A.M.), and Blood Cancer UK Bennett Fellowship (15008) (G.B. and D.G.K.). The authors thank the Protein Production Facility and Bioimaging Facility at the University of Leeds, which are funded by the Royal Society Wolfson Scheme (WL150028) and BBSRC (BB/R000352/1). A.M. is a Scholar of The Leukemia & Lymphoma Society. J.F.R. is funded by a Leeds Anniversary Research Studentship.
Authorship
Contribution: J.F.R., A.J.B., D.G.K., A.M., and E.C. designed the research; J.F.R., A.J.B., F.N., G.B., R.S., H.P., E.L.B., G.A., S.A.B., B.R.J., P.L., A.M., and E.C. performed experiments and collected data; E.J.B. and A.R.G. provided patient samples; J.F.R., A.J.B., P.L., A.M., and E.C. analyzed the data; and J.F.R., A.J.B., F.N., A.M., and E.C. wrote the manuscript.
Conflict-of-interest disclosure: A.M. has consulted for Janssen, PharmaEssentia (steering committee), Constellation (advisory board), and Relay Therapeutics and receives research support from Janssen and Actutate Therapeutics. The remaining authors declare no competing financial interests.
Correspondence: Edwin Chen, School of Life Sciences, University of Westminster, 115 New Cavendish Rd, London, W1W 6UW, United Kingdom; e-mail: e.chen@westminster.ac.uk.

