

Key Points
Bortezomib suppressed the survival of, and the inflammatory cytokine production of, EBV-infected T and NK cells in sCAEBV.
Bortezomib had effects on neoplastic and inflammatory aspects of the sCAEBV xenograft model and can be a therapeutic drug candidate.

Abstract
Systemic chronic active Epstein-Barr virus (EBV; sCAEBV) infection, T- and natural killer (NK)-cell type (sCAEBV), is a fatal disorder accompanied by persisting inflammation harboring clonal proliferation of EBV-infected T or NK cells. Today’s chemotherapy is insufficient to resolve disease activity and to rid infected cells of sCAEBV. The currently established treatment strategy for eradicating infected cells is allogeneic hematopoietic stem cell transplantation. In this study, we focused on the effects of proteasome inhibitor bortezomib on the disease. Bortezomib suppressed survival and induced apoptosis of EBV+ T- or NK-cell lines and peripheral mononuclear cells containing EBV-infected T or NK cells of sCAEBV patients. Bortezomib enhanced binding immunoglobulin protein/78-kDa glucose-regulated protein (Bip/GRP78) expression induced by endoplasmic reticulum stress and activated apoptosis-promoting molecules JNK and p38 in the cell lines. Bortezomib suppressed the activation of survival-promoting molecule NF-κB, which was constitutively activated in EBV+ T- or NK-cell lines. Furthermore, quantitative reverse transcription–polymerase chain reaction demonstrated that bortezomib suppressed messenger RNA expression of proinflammatory cytokines tumor necrosis factor α (TNF-α) and interferon γ (IFN-γ) in EBV+ T or NK cells from the patients. Finally, we examined the effects of bortezomib using xenograft models of sCAEBV generated by IV injection of patients’ cells. The intraperitoneal administration of bortezomib significantly reduced EBV-DNA load in peripheral blood and the infiltration of EBV-infected cells in the models’ livers. Moreover, the serum concentration of TNF-α and IFN-γ decreased after bortezomib treatment to the models. Our findings will be translated into the treatment of sCAEBV not only to reduce the number of tumor cells but also to suppress inflammation.
Introduction
The Epstein-Barr virus (EBV) genome is positive in some T- and natural killer (NK)-cell neoplasms, namely extranodal NK/T-cell lymphoma, nasal type (ENKL), aggressive NK-cell leukemia (ANKL), EBV+ peripheral T-cell lymphoma not otherwise specified (PTCL-NOS), systemic EBV+ T-cell lymphoma in childhood, and chronic active EBV infection (CAEBV). CAEBV was originally reported as a disorder of sustained inflammation similar to infectious mononucleosis.1 Later, it was discovered that CAEBV is accompanied by clonal proliferation of EBV+ T or NK cells.2-5 Based on these findings, the 2017 World Health Organization (WHO) classification defined CAEBV as an EBV+ T- or NK-cell lymphoproliferative disease.6 Hypersensitivity to mosquito bites (HMB) and hydroa vacciniforme–like eruption, 2 disorders in which lesions are limited to skin, were also found to accompany EBV+ T or NK cells with clonal proliferation and reveal similar clinical courses as in CAEBV.5 Therefore, the WHO 2017 classification described 2 subtypes of CAEBV: systemic CAEBV (sCAEBV) accompanied by systemic inflammation and cutaneous CAEBV of HMB or hydroa vacciniforme-like eruption.6
There are 2 aspects of sCAEBV: a lymphoid neoplasm and an inflammatory disorder. EBV infection of T or NK cells renders the cells immortal.7 The infected cells also activate, leading to systemic inflammation with hypercytokinemia.8,9 Furthermore, sCAEBV gradually develops into fatal disorders, such as T- or NK-cell lymphoma or hemophagocytic lymphohistiocytosis.5,10 The only effective treatment strategy to possibly eradicate EBV-infected tumor cells is hematopoietic stem cell transplantation (HSCT),5,11,12 but there are limitations. HSCT is not feasible for all patients, especially for those with poor general condition. In addition, the outcomes of patients with active disease, accompanied by inflammatory symptoms such as fever and liver dysfunction at the time of HSCT, are significantly poorer than those with inactive disease.5,10,12 Therefore, the development of an effective reagent is an urgent need not only to reduce tumor cells but also to regulate the inflammation caused by the disease. We previously determined that NF-κB was constitutively activated through latent membrane protein 1 (LMP1), promoting survival in the EBV+ T or NK cells of CAEBV patients.13 NF-κB was also activated by inflammatory cytokines and lymphocyte antigen stimulation, and then upregulated inflammation-promoting molecules inducing systemic inflammation.14 These findings suggest that NF-κB can be a therapeutic target efficacious for neoplasm and inflammation of sCAEBV.
Proteasome inhibitor bortezomib was originally developed as a reagent for multiple myeloma. Bortezomib induced intracellular accumulation of unfolded proteins by proteasome inhibition, which eventually leads to endoplasmic reticulum (ER) stress.15 Bortezomib suppresses the degradation of IκB, an endogenous inhibitor of NF-κB in the proteasome, and inhibits NF-κB activation.16 It also reduces the activation of some survival-promoting molecules such as Erk and Akt,16,17 which may cause the development of tumors. Bortezomib also activates JNK and p38, generates the upregulation of p53 and the downregulation of BCL-XL, and induces apoptosis of target cells.16,18 These findings prompted studies to examine the efficacy of bortezomib against lymphoid neoplasms other than multiple myeloma. Today, there are reports proving the effects of bortezomib for B-lymphoid malignancies such as mantle cell lymphoma.19 There is another report stating that bortezomib is effective against T- or NK-cell neoplasms including CAEBV.20,21
In this study, we investigated the effects of bortezomib on sCAEBV using not only EBV+ T- or NK-cell lines but also clinical samples obtained from sCAEBV patients. We also investigated the effects on xenograft models established from the transplantation of sCAEBV patients’ EBV+ cells.22 The aim was to examine whether bortezomib is effective against lymphoid neoplasm and inflammatory disorder, the 2 aspects of sCAEBV.
Materials and methods
Cell lines and reagents
SNT8 was derived from the T-cell type of nasal NK/T-cell lymphoma. SNK6 was derived from the NK-cell type of nasal NK/T-cell lymphoma. SNT15 and SNT16 were derived from the T-cell type of CAEBV.23 They were cultured in RPMI-1640 containing 10% fetal calf serum (FCS) and 175 U/mL human interleukin 2 (IL-2) or Artemis Medium-2 (Nihon Techno Service, Ibaraki, Japan). The EBV− T cell line Jurkat was cultured in 10% FCS–RPMI-1640. The EBV− NK-cell line KHYG1 was cultured in 10% FCS–RPMI-1640 containing 175 U/mL human IL-2. IL-2 was purchased from R&D Systems (Minneapolis, MN). Bortezomib was purchased from Millennium Pharmaceuticals, Inc (Cambridge, MA).
Diagnosis of sCAEBV
sCAEBV was diagnosed based on criteria suggested by the Research Group of Measures against Intractable Diseases of the Ministry of Health, Labour and Welfare of Japan, which conform with the definition of CAEBV in the WHO 2017 classification:
elevated EBV-DNA load in peripheral blood (PB; >102.5 copies per microgram of DNA);
EBV infection of T or NK cells in the affected tissues or PB;
systemic inflammatory symptoms (such as fever, lymphadenopathy, liver dysfunction, progressive skin lesions, vasculitis, uveitis) persisting for >3 months; and
exclusion of other possible diagnoses: primary infection of EBV (infectious mononucleosis), autoimmune disease, congenital immunodeficiency, HIV, and other immunodeficiencies or underlying diseases with potential immunosuppression.
The detection of EBV-infected cells in sCAEBV patients
Infected cells were detected and isolated as described previously.24 In brief, PB mononuclear cells (PBMCs) from patients were isolated by density gradient centrifugation using Separate-L (Muto Pure Chemical Co, Ltd, Tokyo, Japan) and sorted into CD19+, CD4+, CD8+, or CD56+ fractions by using antibody-conjugated magnetic beads (Miltenyi Biotec, Bergisch Gladbach, Germany). The EBV-DNA level in each fraction were then measured by real-time reverse transcription–polymerase chain reaction (RT-PCR) using the TaqMan system (Applied Biosystems, Foster City, CA).25 The fraction with the highest titer was determined to contain infected cells. The clonality of infected cells was examined by Southern blotting.
XTT assay
The sodium 3V-[1-(phenylaminocarbonyl)-3,4-tetrazolium]-bis(4-methoxy-6-nitro)-benzene sulfonic acid hydrate (XTT) assay was performed according to the XTT) colorimetric method, using the Cell Proliferation kit II (Roche Molecular Biochemicals, Indianapolis, IN) according to the manufacturer’s instructions. The kit was used to measure cellular metabolic activity as an indicator of viable cells.
Apoptosis detection assay
Apoptosis detection was performed using the TACS Annexin V–FITC apoptosis detection kit (Trevigen, Inc, Gaithersburg, MD) following the manufacturer’s instructions.
Antibodies
For western blotting, phospho–stress-activated protein kinase/JNK (Thr183/Try185), phospho-p38 MAPK (Thr180/Tyr182), poly (ADP-ribose) polymerase-1 (PARP), X-box–binding protein 1s (XBP1s; D2C1F), and LC3A/B were purchased from Cell Signaling Technology, Inc (Danvers, MA). Binding immunoglobulin protein /78-kDa glucose-regulated protein (Bip/GRP78; N20), p53 (DO-1), and heat shock protein 90 α/β (Hsp90α/β; F-8) were purchased from Santa Cruz Biotechnology, Inc. For immunofluorescence staining, we used antibodies against NF-κB p50 (H-119), NF-κB p52 (C-5), NF-κB p65 (F-6), RelB (C-19) (Santa Cruz Biotechnology), and LMP1 (clone S12).26 For immunohistochemistry, we used antibodies against CD3, CD4 (Leica Microsystems Ltd, Newcastle, United Kingdom), CD8 (clone C8/144B), and LMP1 (CS.1-4) (Dako, Glostrup, Denmark).
Western blotting
After washing with phosphate-buffered saline (PBS), cells were lysed in the buffer containing 50 mM Tris–HCl pH 7.5, 5 mM EDTA, 100 mM NaCl, 50 mM NaF, 1 mM Na3VO4, 40 mM β-glycerophosphate, and 1% Triton X-100 as described previously.27 The lysates were centrifuged at 15 000 rpm for 10 minutes, then the supernatant was collected and subjected to western blotting. Chemiluminescent detection was performed using Western Lightning Plus-ECL (Perkin Elmer, Inc, Waltham, MA).
Immunofluorescence staining
The assays were performed as described previously.28 Cells were fixed on slides via immersion in 4% paraformaldehyde for 10 minutes, followed by 3 washes in PBS and incubation with mouse monoclonal anti-LMP1, p52, p50, RelA, or RelB antibodies at room temperature. Next, the slides were treated at room temperature with Alexa Fluor 488 chicken anti-mouse immunoglobulin G (Invitrogen, Carlsbad, CA) to label anti-LMP1, a Cy5-conjugated Affinipure donkey anti-mouse antibody (Jackson ImmunoResearch Laboratories, Inc, West Grove, PA) to label anti-p52 and anti-RelA antibodies, or a phycoerythrin-conjugated goat anti-rabbit antibody (Southern Biotech Associates Inc, Birmingham, AL) to label anti-p50 and anti-RelB antibodies. Nuclei were counterstained with ProLong Gold and 4′,6-diamidino-2-phenylindole (DAPI; Invitrogen, Carlsbad, CA).
Image acquisition
In cytological analysis, we used a confocal microscope (Fluoview FV10i-DOC; Olympus Corporation, Tokyo, Japan). We took photographs with a 60× objective lens (NA 1.35).
RT-PCR
RNA was extracted with ISOGEN II (Nippon Gene Co, Ltd, Tokyo, Japan) from the cell lines and PBMCs of the patients. Complementary DNA reactions were made using Transcriptor Universal cDNA Master (F. Hoffmann-La Roche AG, Basel, Switzerland). Quantitative RT-PCR (qRT-PCR) was performed on Light Cycler 480 (F. Hoffmann-La Roche AG) using TaqMan gene-expression assays (Applied Biosystems). The messenger RNA (mRNA) of viral proteins was measured as previously described.29 We performed qRT-PCR to investigate mRNA of inflammatory cytokine expression, interferon γ (IFN-γ) and tumor necrosis factor α (TNF-α). We used Hs00989291_m1 and Hs01113624_g1 primers for IFN-γ and TNF-α, respectively (Applied Biosystems).
Xenograft model generation and bortezomib treatment
The generation of the NOD/Shi-scid/IL-2Rγnull (NOG) model is as described previously.22,28 In brief, male NOG mice were obtained from the Central Institute for Experimental Animals (Kawasaki, Japan) and maintained under specific pathogen-free conditions. The models were generated by injecting PBMCs from 2 patients (CD4-3 and CD56-1) into 6-week-old mice through the tail vein. The establishment of the models was confirmed by detecting CD4+ or CD56+ cells with a flow cytometer and PCR-based EBV virus genome detection in the PB. Established models were randomly assigned to experimental groups and intraperitoneally injected with 300 μL of PBS or 1.67 mg/kg bortezomib in 300 μL of PBS twice a week, as described previously.30 The EBV-DNA load in the PB was monitored weekly. Tribromoethanol anesthesia was administered IV to minimize suffering. After the experiment, mice were euthanized via CO2 inhalation and subjected to analyses.
Immunohistochemistry
Paraffin-embedded formalin-fixed tissue sections were deparaffinized, and heat-based antigen retrieval was performed in 0.1 mol/L citrate buffer (pH 6.0). Endogenous peroxidase activity was inhibited using hydrogen peroxide. The detection system was a streptavidin-biotin-peroxidase complex technique (ABC Kits; Vector Laboratories, Burlingame, CA) with diaminobenzidine (Vector Laboratories) as the chromogen. In situ hybridization (ISH) of EBV-encoded small RNA (EBER) was performed to detect EBV in tissue sections (by EBV [EBER] PNA Probe/Dako Fluorescein; Agilent Technologies, Inc, Santa Clara, CA) and a second antibody (Molecular Probes Fluorescein; Thermo Fisher Scientific Inc, Waltham, MA).
Quantification of cytokines
The cytokine concentrations in the serum of the xenograft models were determined by enzyme-linked immunosorbent assay using the Quantikine ELISA kit (R&D Systems) following the manufacturer’s instructions.
Statistical analysis
Statistical analyses were performed using GraphPad Prism 5 (GraphPad Software, San Diego, CA). The significance was calculated using the Student t test. For survival studies, we used the log-rank test.
Ethics statement
The study complied with the Declaration of Helsinki and was approved by the ethical committee of Tokyo Medical and Dental University (TMDU). Written informed consent was obtained from each patient. The experiments with NOG mice are in accordance with the Guidelines for Animal Experimentation of the Japanese Association for Laboratory Animal Science, as well as ARRIVE guidelines.31 The experiments were approved by the Institutional Animal Care and Use Committees of TMDU (A2018-293C5) and the National Research Institute for Child Health and Development (A2015-001-C04, A2015-002-C04).
Results
Bortezomib inhibits survival and induces apoptosis in EBV+ T- or NK-cell lines
First, we investigated the effects of bortezomib on EBV+ T- or NK-cell lines. The numbers of living cells of EBV+ T-cell lines SNT8, SNT15, and SNT16, which were treated with 1 to 10 nM bortezomib, were significantly lower than those of EBV− cell line Jurkat treated with the same concentration of bortezomib (Figure 1A). Bortezomib (1-50 nM) also suppressed more significant numbers of living cells of EBV+ NK-cell line SNK6 compared with the numbers of EBV− NK-cell line KHYG1 (Figure 1A). The Annexin V assay showed that treatment with 10 nM bortezomib, the optimal concentration for clinical use, remarkably induced apoptosis in the EBV+ cell lines (Figure 1B). These results indicate that bortezomib suppressed survival and induced apoptosis in EBV+ T- or NK-cell lines. It has been reported that bortezomib induced the lytic gene expression of EBV, leading to the replication of the virus.32 The expression of viral proteins was examined. BZLF1 and gp220/350, which were indispensable for the replication of the virus, were not induced and EBV-DNA load did not increase (supplemental Figure 1). LMP1 protein expression was 72% to 83% in all cell lines and did not significantly increase after treatment (supplemental Figure 2).
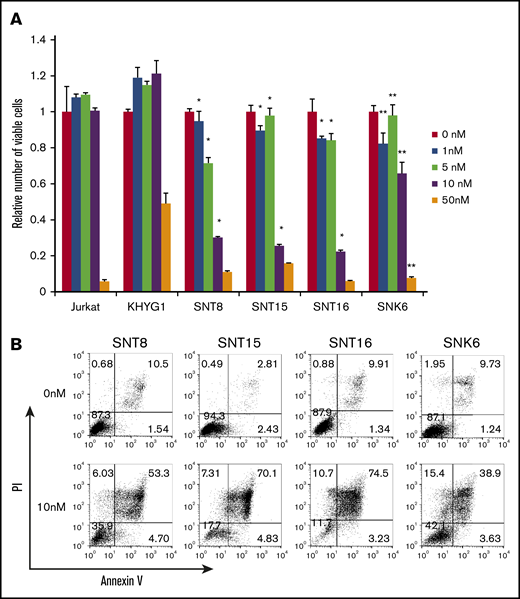
The effects of bortezomib on survival of EBV+T- or NK-cell lines. (A) EBV+ T- or NK-cell lines, SNT8, SNT15, SNT16, and SNK6 cells were treated with bortezomib for 48 hours, and the viable cell number was estimated using the XTT assay and expressed in arbitrary units. EBV− T- or NK-cell lines, Jurkat, and KHYG1 cells, respectively, were used as negative controls. The data represent the mean plus or minus standard deviation (SD) of 3 independent experiments. The number of viable cells of SNT8, SNT15, SNT16, and SNK6 were compared with those of EBV− cell lines, Jurkat (*) and KHYG1 (**), treated with the same concentration of bortezomib (P < .05). (B) SNT8, SNT15, SNT16, and SNK6 cells were treated with bortezomib for 48 hours and used for the assay. Cells were stained with Annexin V and propidium iodide (PI), then analyzed by flow cytometry.
The effects of bortezomib on survival of EBV+T- or NK-cell lines. (A) EBV+ T- or NK-cell lines, SNT8, SNT15, SNT16, and SNK6 cells were treated with bortezomib for 48 hours, and the viable cell number was estimated using the XTT assay and expressed in arbitrary units. EBV− T- or NK-cell lines, Jurkat, and KHYG1 cells, respectively, were used as negative controls. The data represent the mean plus or minus standard deviation (SD) of 3 independent experiments. The number of viable cells of SNT8, SNT15, SNT16, and SNK6 were compared with those of EBV− cell lines, Jurkat (*) and KHYG1 (**), treated with the same concentration of bortezomib (P < .05). (B) SNT8, SNT15, SNT16, and SNK6 cells were treated with bortezomib for 48 hours and used for the assay. Cells were stained with Annexin V and propidium iodide (PI), then analyzed by flow cytometry.
Bortezomib induces ER stress–related molecule expression and regulates survival-related molecules in EBV+ T- or NK-cell lines
It has been reported that bortezomib induced intracellular accumulation of unfolded proteins by proteasome inhibition, which eventually led to ER stress.15 We then examined the molecules associated with ER stress, Bip/GRP78 and XBP1s. A chaperone protein, Bip/GRP78 is induced by ER stress and contributes to the degradation of unfolded proteins. As shown in Figure 2A, the expression of Bip was upregulated by bortezomib in all examined cell lines. These findings indicate that bortezomib induced ER stress in the cell lines. XBP1 is a protein induced by ER stress downstream of Bip/GRP78 via inositol-requiring enzyme 1 alpha (IRE1α) and mediates apoptosis-promoting intracellular signaling.33 However, as shown in supplemental Figure 3, bortezomib-induced XBP1s expression was detected only in SNT8 and was not parallel to Bip/GRP78 expression in the other cells. Instead, p38 and JNK, molecules that mediate apoptosis-promoting signaling downstream of Bip/GRP78 other than XBP1, were activated by bortezomib in all examined cell lines. Bortezomib also induced p53, the expression of which was upregulated by JNK (Figure 2B). The cleavage of PARP was also detected (Figure 2B). These results indicate that bortezomib induced ER stress, resulting in apoptosis in EBV+ T- or NK-cell lines. Bortezomib induced the expression of LC3, which mediates autophagy in all examined cells (Figure 2C).
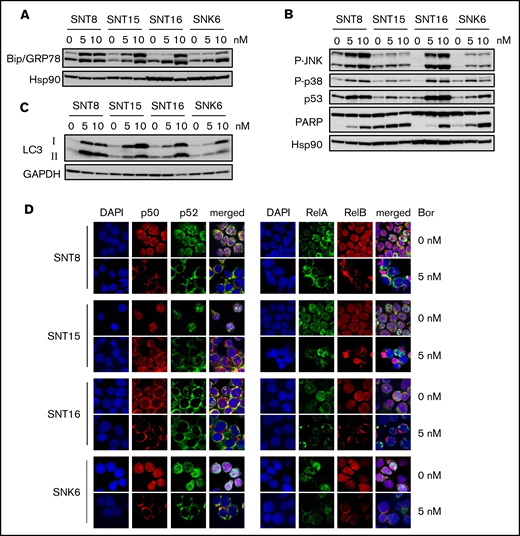
The effects of bortezomib on intracellular signaling–mediating molecules in EBV+T- or NK-cell lines. (A-C) SNT8, SNT15, SNT16, and SNK6 cells were treated with bortezomib for 24 hours and subjected to immune blotting. Hsp90 or glyceraldehyde-3-phosphate dehydrogenase (GAPDH) served as a loading control. (D) SNT8, SNT15, SNT16, and SNK6 cells were treated with bortezomib for 24 hours. The expression and the localization of NF-κB proteins were examined by immune-fluorescent staining with anti-p50, p52, RelA, and RelB antibodies. DAPI was for nuclei; original magnification ×60. The cells were analyzed by confocal microscopy.
The effects of bortezomib on intracellular signaling–mediating molecules in EBV+T- or NK-cell lines. (A-C) SNT8, SNT15, SNT16, and SNK6 cells were treated with bortezomib for 24 hours and subjected to immune blotting. Hsp90 or glyceraldehyde-3-phosphate dehydrogenase (GAPDH) served as a loading control. (D) SNT8, SNT15, SNT16, and SNK6 cells were treated with bortezomib for 24 hours. The expression and the localization of NF-κB proteins were examined by immune-fluorescent staining with anti-p50, p52, RelA, and RelB antibodies. DAPI was for nuclei; original magnification ×60. The cells were analyzed by confocal microscopy.
We subsequently investigated the effects of bortezomib on NF-κB activation. Immune-fluorescent staining demonstrated that treatment with 5 nM bortezomib inhibited the nuclear translocation of p50, p52, RelA, and RelB in EBV+ cell lines (Figure 2D). These results indicate that bortezomib suppressed survival-promoting molecules NF-κB and activated apoptosis-inducing molecules in EBV+ T- or NK-cell lines.
Bortezomib inhibits the survival and the expression of the inflammatory cytokines of PBMCs obtained from sCAEBV patients
Next, we examined the effects of bortezomib on the samples from sCAEBV patients. sCAEBV was diagnosed according to the criteria shown in “Materials and methods.” In this study, we investigated 10 sCAEBV patients between 18 and 64 years of age; 2 were male and 8 were female. Six were T-cell type and 3 were NK-cell type: CD4 type (n = 3), CD8 type (n = 3), and CD56 type (n = 3). One patient had both EBV-infected T (CD4) and NK (CD56) cells. Pathological diagnosis of CAEBV was made in 7 patients. In CD4-2, CD56-2, and CD56-3, diagnosis of CAEBV was made by clinical findings and detection of EBV-infected T or NK cells using unfixed PB as shown in “Materials and methods.” The patients with overt EBV+ lymphomas, such as ENKL, ANKL, and PTCL-NOS, were ruled out.
The clinical findings, the phenotype of the infected cells, and their EBV-DNA load are shown in Table 1. An XTT assay revealed that bortezomib (0.5-50 nM) suppressed the survival of PBMCs that contained EBV+ T or NK cells from the 9 sCAEBV patients, but did not affect PBMCs of healthy donors (Figure 3A).
The characteristics of the patients
| Case | Sex | Age, y | Infected cell | Clonality of the infected cells | Clinical findings | EBV-DNA load of each fraction of PBMCs, copies/μg DNA | Pathological diagnosis | The rate of the EBV-infected cell fractions in PBMCs, % | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Total PBMCs | CD4+ cells | CD8+ cells | CD56+ cells | Others | ||||||||
| CD4-1 | F | 25 | CD4 | Monoclonal | Fever, HMB | 7.0 × 104 | 2.2 × 105 | ND | ND | ND | CAEBV | 71.2 |
| CD4-2 | F | 62 | CD4 | Monoclonal | Fever, LD | 3.2 × 104 | 4.6 × 105 | ND | ND | ND | ND* | 23.5 |
| CD4-3 | M | 33 | CD4 | Monoclonal | Fever | 9.2 × 103 | 1.0 × 105 | ND | ND | ND | CAEBV | 54.2 |
| CD8-1 | F | 21 | CD8 | Monoclonal | Fever, LD | 1.8 × 103 | ND | 6.0 × 102 | ND | ND | CAEBV | 50.0 |
| CD8-2 | F | 64 | CD8 | Monoclonal | Fever, LD | 2.6 × 105 | ND | 1.2 × 106 | ND | ND | CAEBV | 51.0 |
| CD8-3 | F | 64 | CD8 | Monoclonal | Fever, LD | 4.7 × 104 | ND | 1.7 × 104 | ND | ND | CAEBV | 51.1 |
| CD56-1 | F | 18 | CD56 | Monoclonal | Fever, HMB | 5.2 × 104 | ND | ND | 1.1 × 106 | ND | CAEBV | 3.8 |
| CD56-2 | F | 48 | CD56 | Monoclonal | Fever, LD | 8.6 × 104 | ND | ND | 1.0 × 105 | ND | ND* | 10.0 |
| CD56-3 | M | 23 | CD56 | Monoclonal | Fever, colitis | 2.9 × 103 | ND | ND | 2.1 × 104 | ND | ND* | 4.2 |
| CD4/56-1 | F | 18 | CD4/56 | Monoclonal | Fever, LD | 6.0 × 104 | 1.7 × 106 | ND | 7.1 × 106 | ND | CAEBV | 35.2 (CD4), 2.3 (CD56) |
| Case | Sex | Age, y | Infected cell | Clonality of the infected cells | Clinical findings | EBV-DNA load of each fraction of PBMCs, copies/μg DNA | Pathological diagnosis | The rate of the EBV-infected cell fractions in PBMCs, % | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Total PBMCs | CD4+ cells | CD8+ cells | CD56+ cells | Others | ||||||||
| CD4-1 | F | 25 | CD4 | Monoclonal | Fever, HMB | 7.0 × 104 | 2.2 × 105 | ND | ND | ND | CAEBV | 71.2 |
| CD4-2 | F | 62 | CD4 | Monoclonal | Fever, LD | 3.2 × 104 | 4.6 × 105 | ND | ND | ND | ND* | 23.5 |
| CD4-3 | M | 33 | CD4 | Monoclonal | Fever | 9.2 × 103 | 1.0 × 105 | ND | ND | ND | CAEBV | 54.2 |
| CD8-1 | F | 21 | CD8 | Monoclonal | Fever, LD | 1.8 × 103 | ND | 6.0 × 102 | ND | ND | CAEBV | 50.0 |
| CD8-2 | F | 64 | CD8 | Monoclonal | Fever, LD | 2.6 × 105 | ND | 1.2 × 106 | ND | ND | CAEBV | 51.0 |
| CD8-3 | F | 64 | CD8 | Monoclonal | Fever, LD | 4.7 × 104 | ND | 1.7 × 104 | ND | ND | CAEBV | 51.1 |
| CD56-1 | F | 18 | CD56 | Monoclonal | Fever, HMB | 5.2 × 104 | ND | ND | 1.1 × 106 | ND | CAEBV | 3.8 |
| CD56-2 | F | 48 | CD56 | Monoclonal | Fever, LD | 8.6 × 104 | ND | ND | 1.0 × 105 | ND | ND* | 10.0 |
| CD56-3 | M | 23 | CD56 | Monoclonal | Fever, colitis | 2.9 × 103 | ND | ND | 2.1 × 104 | ND | ND* | 4.2 |
| CD4/56-1 | F | 18 | CD4/56 | Monoclonal | Fever, LD | 6.0 × 104 | 1.7 × 106 | ND | 7.1 × 106 | ND | CAEBV | 35.2 (CD4), 2.3 (CD56) |
F, female; HMB, hypersensitivity to mosquito bites; LD, liver dysfunction; M, male; ND, not detected.
The diagnosis of CAEBV was made by clinical findings and detecting EBV-infected T or NK cells using unfixed PB as shown in “Materials and methods.”
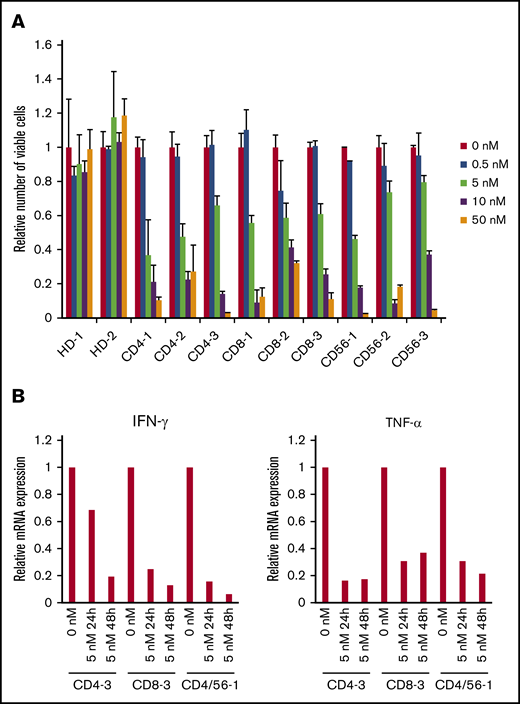
The effects of bortezomib on EBV+T or NK cells from systemic CAEBV patients. (A) The PBMCs from 9 systemic CAEBV patients and 2 healthy donors were treated with bortezomib in the presence of IL-2 for 48 hours, and the viable cell number was estimated using the XTT assay and expressed in arbitrary units. The data represent the mean plus or minus SD of 3 independent experiments. (B) PBMCs from systemic CAEBV patients (CD4-3, CD8-3, and CD4/CD56-1) were treated with 5 nM bortezomib for 24 hours and 48 hours. The mRNA expression of IFN-γ and TNF-α in PBMCs was examined by qRT-PCR.
The effects of bortezomib on EBV+T or NK cells from systemic CAEBV patients. (A) The PBMCs from 9 systemic CAEBV patients and 2 healthy donors were treated with bortezomib in the presence of IL-2 for 48 hours, and the viable cell number was estimated using the XTT assay and expressed in arbitrary units. The data represent the mean plus or minus SD of 3 independent experiments. (B) PBMCs from systemic CAEBV patients (CD4-3, CD8-3, and CD4/CD56-1) were treated with 5 nM bortezomib for 24 hours and 48 hours. The mRNA expression of IFN-γ and TNF-α in PBMCs was examined by qRT-PCR.
In sCAEBV patients, EBV+ T or NK cells produced inflammatory cytokines IFN-γ and TNF-α. Their serum concentrations increased and reflected the status of the disease.9 We investigated mRNA of IFN-γ and TNF-α expression in the PBMCs of sCAEBV patients by qRT-PCR. We examined 3 patients (CD4-3, CD8-3, and CD4/56-1) in this experiment. Bortezomib decreased the inflammatory cytokine expression in a time-dependent manner (Figure 3B). Bortezomib did not show significant effects on IFN-γ and TNF-α expression in EBV+ T- or NK-cell lines (data not shown).
Bortezomib has effects on an sCAEBV xenograft model
Finally, we investigated the effects of bortezomib in xenograft models of sCAEBV. The models were generated by transplanting PBMCs from 2 CAEBV patients, CD4-3 and CD56-1, to NOG mice as described in “Materials and methods.” After transplantation, engraftments in the models were determined by detecting the EBV genome in the PBMCs. Most of the cells from PB and the spleen were CD4 or CD56+. CD19+ cells were either not detected or extremely low in number (supplemental Figure 4A-B). These results indicate that the infected cells were enriched in the PB and the spleen of the models. Histopathological images of the liver are shown in supplemental Figure 4C-D. Infiltrating EBER+ cells were detected in the periportal regions. CD4+ and CD8+ cells were detected. CD3ε+ cells were also confirmed substantially, and their numbers were larger than those of CD4+ and CD8+ cells in NK-cell type mice (supplemental Figure 4D); some could have been NK cells.
PBS and bortezomib were injected intraperitoneally twice a week to the xenograft models. Twenty-two mice, 11 for PBS and 11 for bortezomib, were evaluated through weekly quantification. Bortezomib treatment significantly reduced the EBV-DNA load in PB relative to PBS-treated mice (Figure 4A). However, 3 weeks from treatment, EBV-DNA tended to increase again. The results from CD4 cell-type models and CD56 cell-type models were analyzed separately as shown in supplemental Figure 5. In the CD56 cell-type model, EBV-DNA of the bortezomib-treated mice decreased statistically in comparison with the PBS-treated mice (supplemental Figure 5C), whereas this difference was observed in the CD4-cell type model but was not significant (supplemental Figure 5A). There was no significant difference in weight between the PBS-treated and the bortezomib-treated mice (Figure 4B; supplemental Figure 5B,D). We investigated the serum concentrations of IFN-γ and TNF-α in CD4+ cell–transplanted mice. The serum concentrations decreased after bortezomib treatment (Figure 4C-D). Histopathological examination of the PBS- and the bortezomib-treated mice is shown in Figure 4E. CD4+ cell–transplanted mice were examined and the representative images are shown. The infiltrating cell number notably decreased in the periportal regions in the bortezomib-treated mice 3 weeks after treatment initiation. The number of EBER+ cells significantly decreased in bortezomib-treated mice tissue. The survival curves of the PBS-treated mice and the bortezomib-treated mice are shown in Figure 4F, and those of the CD4 cell type and the CD56 cell type are shown in supplemental Figure 5E-F. We did not observe any statistically significant difference individually or as a whole. These results indicate that bortezomib had effects on both neoplastic and inflammatory aspects of CAEBV. We examined the expression of the lytic genes of EBV: BZLF1 and gp350 in PB of the models. As shown in supplemental Table 1, bortezomib did not induce the expression of mRNA for BZLF1 and gp220/350 in the models. We concluded that bortezomib did not induce the reactivation of EBV in CAEBV patients.

The effects of bortezomib on the systemic CAEBV xenograft model. The xenograft model of systemic CAEBV was generated using NOG mice. Mice were injected intraperitoneally with 300 μL of PBS, or 1.67 mg/kg bortezomib in 300 μL of PBS twice a week for 3 weeks. (A-B) PBS and bortezomib were administered to 11 mice for each. The EBV-DNA load of PB and the body weight were monitored weekly. *P < .05. (C-D) The concentration of cytokines in the serum was measured every week among 3 mice established by injecting the PBMCs of the CD4-3 patient. The data represent the mean plus or minus SD of each independent experiments. (E) Representative images of hematoxylin/eosin (HE) staining and EBER ISH are shown after 3 weeks of treatment with bortezomib or PBS in the mice established by injecting PBMCs of the CD4-3 patient. Original magnification ×400. (F) The survival curve of the bortezomib-treated mice (n = 40: 20 CD4 cell type, 20 CD56 cell types) and the PBS-treated mice (n = 40: 20 CD4 cell type, 20 CD56 cell types) was analyzed by the log-rank test.
The effects of bortezomib on the systemic CAEBV xenograft model. The xenograft model of systemic CAEBV was generated using NOG mice. Mice were injected intraperitoneally with 300 μL of PBS, or 1.67 mg/kg bortezomib in 300 μL of PBS twice a week for 3 weeks. (A-B) PBS and bortezomib were administered to 11 mice for each. The EBV-DNA load of PB and the body weight were monitored weekly. *P < .05. (C-D) The concentration of cytokines in the serum was measured every week among 3 mice established by injecting the PBMCs of the CD4-3 patient. The data represent the mean plus or minus SD of each independent experiments. (E) Representative images of hematoxylin/eosin (HE) staining and EBER ISH are shown after 3 weeks of treatment with bortezomib or PBS in the mice established by injecting PBMCs of the CD4-3 patient. Original magnification ×400. (F) The survival curve of the bortezomib-treated mice (n = 40: 20 CD4 cell type, 20 CD56 cell types) and the PBS-treated mice (n = 40: 20 CD4 cell type, 20 CD56 cell types) was analyzed by the log-rank test.
Discussion
We discovered that bortezomib suppresses the survival of EBV+ T or NK cells and the production of inflammatory cytokines. There are other reports pointing out the effectiveness of bortezomib against the EBV+ T- or NK-cell neoplasms.21,34 The significance of our study is that bortezomib presented not only its antitumor effect but also an anti-inflammatory effect.
According to the previous report, the optimal concentration of bortezomib in PB is considered to be ∼10 nM after administration in clinical use.35 Our results indicate that the concentration of 10 nM is high enough to induce apoptosis as well as to suppress the cytokine production of EBV+ T or NK cells, whereas the same concentration dosage did not show significant effects on lymphocytes of healthy donors. Additionally, bortezomib did not increase either the lytic protein expression or EBV-DNA in vitro. Iwata et al reported that bortezomib enhanced BZLF1 and gp350/220 expression in EBV+ T-cell lines.21 However, they did not explain the impact on EBV-DNA load. Besides, they used 1 μM bortezomib, which is a remarkably high concentration compared with the concentration we observed in the bortezomib-treated patients. These results indicate that bortezomib can be an effective reagent for sCAEBV without replication of the virus.
sCAEBV is a bifaceted disease of inflammatory and neoplastic elements. In this study, bortezomib dramatically decreased the mRNA level of inflammatory cytokines of TNF-α and IFN-γ in patient-derived EBV+ T or NK cells. The assay using xenograft models showed that bortezomib actually reduced the concentration of TNF-α and IFN-γ in the serum. These results indicate that bortezomib had effects on inflammatory aspects of sCAEBV. The inflammation can be a cause of multiorgan failure and may ultimately become hemophagocytic lymphohistiocytosis. Furthermore, 3 previous reports proved that sCAEBV patients with active disease, which was accompanied by systemic inflammation such as fever, liver dysfunction, vasculitis, progressive skin lesions, or uveitis at transplantation, revealed significantly inferior outcomes than those with inactive diseases.5,10,12 Our results signify that bortezomib may possibly remove disease activity and improve outcomes after HSCT.
The other face of sCAEBV is neoplasm. During the course of clonal proliferation, EBV-infected cells develop chemotherapy-resistant T- or NK-cell lymphoma.36,37 Although there was no statistical significance, bortezomib tended to suppress the survival of sCAEBV xenograft models (Figure 4F). Bortezomib also reduced the EBV-DNA loads in PB and infiltrated EBV+ cells in the organs of xenograft mice. Therefore, we assumed that bortezomib revealed antineoplastic effects on sCAEBV in vivo. On the other hand, the EBV-DNA load, suppressed after bortezomib treatment, reverted to its original level after 3 weeks. The suppression was only temporary and the scarcity of the infected cells in the pathological specimen was different from what we observed in PB. There are possibly 2 reasons for this phenomenon. First, the bortezomib-resistant fractions of EBV+ cells could have survived and have proliferated in PB. Second, the virus including its particle could have increased in PB. As shown in supplemental Table 1, the viral proteins, which expressed prior to the replication of the virus, were not detected after bortezomib treatment. Likewise, we did not observe the increase of the viral load by in vitro assay (supplemental Figure 1). These results indicate that bortezomib did not induce viral replication. Contrarily, some reports have mentioned that bortezomib enhanced the expression of lytic proteins, increasing the viral copy number of EBV although these results were from assays using EBV+ B-cell lines.38,39 Bollard and Cohen suggested combining bortezomib and an antiviral drug, ganciclovir, for T-cell–type sCAEBV.34 Ganciclovir was phosphorylated and activated by EBV protein kinase induced by bortezomib treatment.40 They observed a marked reduction of EBV-DNA load in PB after this combination treatment. Further examinations should clarify the effects of bortezomib and whether or not bortezomib induces lytic infection of EBV in sCAEBV in vivo. Recently, a new flow cytometry technique combining surface marker staining with in situ hybridization for the EBER RNAs has been developed.41,42 Sequential examination of PB using this method, without dissecting mice, shall enable us to analyze the effects of bortezomib on EBV-infected cells in vivo in more detail.
We identified the bortezomib-induced molecular pathways in EBV+ T or NK cells. Bortezomib suppressed degradation of the signaling molecules via proteasome suppression; then, as a result, it induced the accumulation of ubiquitinated proteins leading to the promotion of ER stresses.15 In our study, bortezomib upregulated the expression of Bip/GRP78, which may have been induced by ER stress (Figure 2A). Thus, we determined that proapoptotic pathways were activated by bortezomib-induced ER stress in EBV+ T or NK cells. Although the expression of XBP1s was not induced by bortezomib in all cell lines except for one, the activation of p38 and JNK, which can be activated downstream of ER stress, was observed in all examined cells.39,43 It has been reported that p38 induces apoptosis by upregulating proapoptotic gene NOXA.18 JNK induces p53 expression by the inhibition of its degradation and by the upregulation of expression in various cancer cells.44,45 Consequently, p38 and JNK may have played main roles in these phenomena. Previously, we found that NF-κB was constitutively activated and contributed to the survival of EBV-infected T or NK cells in sCAEBV.13 Moreover, in EBV-infected cells, NF-κB activates STAT3, which mediates molecular signaling downstream of inflammatory cytokine receptors.46 NF-κB may be a molecule playing a key role for bortezomib’s antineoplastic and anti-inflammatory effects. The enhancement of LC3 suggests that bortezomib can induce autophagy in EBV+ T or NK cells. We plan further investigations.
Bortezomib may have effects not only on tumor cells but also on stromal cells.47 In CAEBV, various cells such as EBV− lymphocytes and histiocytes are detected in the lesions including PB.7,36,41 EBV+ cells also infiltrate peripheral vessels especially the sinusoidal lesions of the liver.36 These cells, lymphocytes, histiocytes, and vascular endothelial cells may have influences in the development of sCAEBV by both direct, cell-to-cell, and indirect factor-mediating effects. In this study, bortezomib suppressed the proliferation of PBMCs of patients, even in the cases of low-ratio EBV+ T or NK cells. Bortezomib has the potential to be effective for these nonneoplastic bystander cells in the lesions.
In conclusion, we believe that bortezomib may be an attractive reagent for sCAEBV, suppressing not only neoplastic cells but also disease activity. To further prove its efficacy, we are preparing for another examination to verify the results, this time with a larger number of model mice and a clinical study to validate the effects of bortezomib on sCAEBV.
For original data, please contact the corresponding author, Ayako Arai, at ara.hema@tmd.ac.jp.
Acknowledgments
The authors are grateful to Miori Inoue and Shinya Ayabe for excellent technical assistance. The authors also give special thanks to Ayako Komoto, an assistant financed by funding from the Japan Agency for Medical Research and Development (AMED), for excellent editorial support to the authors during the preparation of this manuscript.
This work was supported by a grant from the Practical Research Project for Rare/Intractable Diseases (18ek0109334h0001, 19ek0109334h0002, 20ek0109334h0003) from AMED.
Authorship
Contribution: M.Y. and A.A. designed the research, performed the experiments, analyzed the data, and wrote the draft; H.S., K.-I.I., F.K., A.O., and M.K. performed the experiments and analyzed the data; M.Y., N.S., M.N., and S.F. analyzed the data; A.A. and N.S. collected the samples: and all authors contributed to the modification of the draft and approved the final submission.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Ayako Arai, Division of Hematology and Oncology, Department of Internal Medicine, School of Medicine, St. Marianna University, 2-16-1 Sugao, Miyamae-ku, Kawasaki, Kanagawa, Japan; e-mail: ara.hema@tmd.ac.jp.

