

TO THE EDITOR:
Advanced systemic mastocytosis (advSM) is a clonal stem cell neoplasm that includes aggressive SM (ASM), SM with an associated hematologic neoplasm (AHN), and mast cell leukemia.1-3 The median survival is 41 months for ASM, 24 months for SM-AHN, and <6 to 18 months for mast cell leukemia.4-6 KIT D816V mutation can be detected in >90% of SM patients.7 Interestingly, multilineage involvement of KIT D816V and multimutations in other genes such as SRSF2, ASXL1, and RUNX1 are frequently detected in advSM.8 Some of the above-mentioned mutations were found to precede the KIT D816V mutation, indicating that the KIT mutation is a phenotypic mutation in SM.9 The presence and number of several mutated genes such as the SRSF2-ASXL1-RUNX1 panel are associated with worse prognosis in advSM.10-12
Recently, the multikinase inhibitor midostaurin was approved for the treatment of advSM on the basis of its clinical activity in 116 adults with advSM.13 Currently, both primary and acquired resistance to midostaurin are clinical challenges in advSM. The complexity and dynamics of mutational profiles in midostaurin-treated advSM have been studied by using serial next-generation sequencing, and acquisition of additional mutations or increasing variant allele frequency (VAF) in KRAS/NRAS, RUNX1, IDH2, or NPM1 were associated with progression.14 However, the changes in clonal architecture under the selection pressure of midostaurin in advSM are still elusive. In contrast to traditional bulk DNA sequencing (DNA-seq), genomic analysis at single-cell resolution may provide a better opportunity to resolve clonal architecture in advSM. Here, we report the clonal evolution and heterogeneity in a case of ASM associated with chronic myelomonocytic leukemia (CMML) being treated with midostaurin and azacitidine at the single-cell level.
A 73-year-old woman presented with night sweats, weight loss of 13 kg, epigastric pain, and right chest wall pain, which she had experienced for 1 year. Her hemogram revealed leukocytosis, monocytosis, and leukoerythroblastosis. A bone marrow (BM) examination was performed, and it revealed a significant increase in abnormal mast cell infiltration and aggregation (∼40%) in January 2018. She tested positive for the KIT D816V mutation, and plasma tryptase was >200 ng/mL. Her disease progressed to ASM with intermittent abdominal pain related to significant spleen involvement, ascites, sclerotic and osteolytic bone lesions, anemia, and thrombocytopenia in November 2018. She was treated with peginterferon alfa-2a plus methylprednisolone for 3 weeks, and she then discontinued treatment because of intolerance in January 2019. Starting in March 2019, she was given midostaurin up to 100 mg/day (maintained at 50 mg/day because of intolerance) to treat her advSM. Her symptoms improved gradually, and partial response was achieved after treatment. However, progressive monocytosis (absolute monocyte count of 31 372 cells per µL) was found in June 2019. Another BM examination revealed a slight decrease of abnormal mast cell aggregation to ∼20% to 30% with increased monocytes, indicating a diagnosis of ASM associated with CMML-1. In addition to midostaurin, azacitidine at 75 mg/m2 for 7 days once every 4 weeks was started in July 2019 to control her CMML-1. Complete hematologic remission was rapidly achieved after the first cycle of azacitidine, and her disease was well controlled with midostaurin plus azacitidine for 8 months. Nevertheless, she experienced rapid disease progression in March 2020, and then died as a result of severe hypercalcemia and sepsis. To investigate clonal evolution and to explore the molecular markers of drugs resistance, we analyzed peripheral blood mononuclear cells (PBMCs) and unsorted bulk BM cells at 3 distinct time points: before treatment (PB1), after midostaurin treatment (BM), and at relapse (PB2) using conventional bulk sequencing (bulk-seq) and single-cell DNA-seq.
Genomic testing was approved by the MacKay Memorial Hospital Institutional Review Board, and the patient had provided written informed consent. PBMCs and unsorted BM cells were used for testing. Single-cell targeted DNA-seq was performed using the Tapestri acute myeloid leukemia panel (Mission Bio, San Francisco, CA). The Archer VariantPlex Myeloid panel (ArcherDX, Boulder, CO) was used for bulk targeted DNA-seq. The detailed description of mutation analysis can be found in the supplemental Materials.
In the bulk-seq data, we identified heterozygous somatic RUNX1 R204Q, SRSF2 P95H, ASXL1 R693*, KIT D816V, and EZH2 V696E mutations in the patient before any treatment was given (Table 1). After treatment with midostaurin, KIT and EZH2 mutations became undetectable, but secondary acquired TET2 P851Lfs*22 and NRAS G12D mutations were detected. At the time of relapse after treatment with midostaurin plus azacitidine, significant decrease of the VAFs in TET2 and NRAS mutations, re-emergence of the KIT and EZH2 mutations, and a second RUNX1 R162S mutation were detected. Notably, SRSF2, ASXL1, and RUNX1 mutations were detected at all 3 time points with similar VAFs, indicating that they were likely co-mutations arising early in the hematopoietic stem and progenitor cells (Figure 1A).
Mutations detected by bulk DNA next-generation sequencing in a patient with advSM
| Variant | VAF, % | ||
|---|---|---|---|
| PB1 | BM | PB2 | |
| RUNX1 R204Q | 34.82 | 46.15 | 20.94 |
| SRSF2 P95H | 34.23 | 43.19 | 20.06 |
| ASXL1 R693* | 35.71 | 45.01 | 20.93 |
| KIT D816V | 30.25 | 0.00 | 12.27 |
| EZH2 V696E | 33.08 | 0.00 | 13.88 |
| TET2 P851Lfs*22 | 0.00 | 44.03 | 6.55 |
| NRAS G12D | 0.00 | 40.87 | 0.00 |
| RUNX1 R162S | 0.00 | 0.00 | 9.97 |
| Variant | VAF, % | ||
|---|---|---|---|
| PB1 | BM | PB2 | |
| RUNX1 R204Q | 34.82 | 46.15 | 20.94 |
| SRSF2 P95H | 34.23 | 43.19 | 20.06 |
| ASXL1 R693* | 35.71 | 45.01 | 20.93 |
| KIT D816V | 30.25 | 0.00 | 12.27 |
| EZH2 V696E | 33.08 | 0.00 | 13.88 |
| TET2 P851Lfs*22 | 0.00 | 44.03 | 6.55 |
| NRAS G12D | 0.00 | 40.87 | 0.00 |
| RUNX1 R162S | 0.00 | 0.00 | 9.97 |
PB1, PBMC sample 7 months before treatment; BM, bulk BM sample 3 months after midostaurin treatment; PB2, PBMC sample at relapse after combination treatment with midostaurin and azacitidine for 8 months.
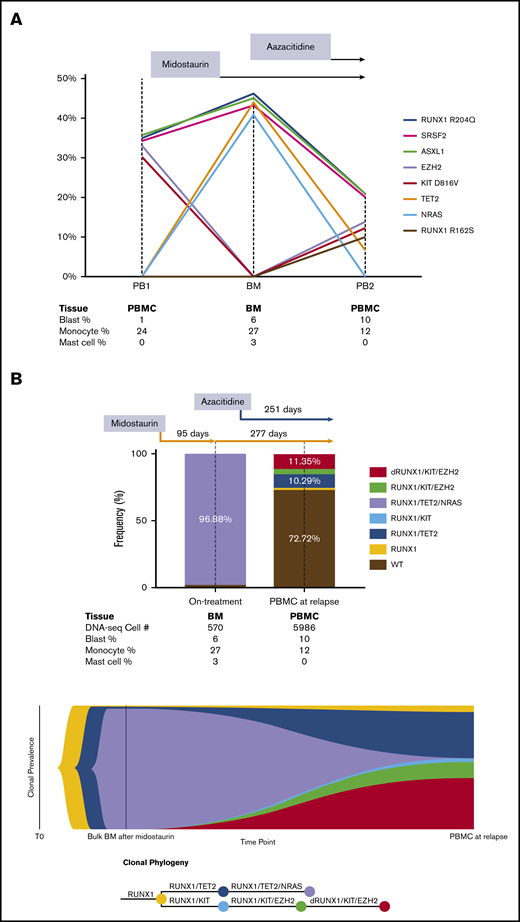
Clonal evolution by serial bulk DNA next-generation sequencing and single-cell DNA-seq in a patient with advSM. (A) Clonal evolution in response to midostaurin and azacitidine by serial bulk DNA-seq. Founding ASXL1, SRSF2, and RUNX1 mutations persisted during midostaurin and azacitidine treatment. KIT and EZH2 mutation burden were initially high at the PB1 time point and were significantly diminished after midostaurin treatment. At the BM time point, allele frequency of NRAS and TET2 mutations were initially high, but they later decreased as a result of azacitidine treatment. KIT and EZH2 mutations, followed by disease relapse, recurred with an additional new mutation site at RUNX1 R162S. The results were projected into a trend line with VAF shown on the Y-axis and collected samples listed below the graph. (B) Clonal evolution in response to treatment with midostaurin and azacitidine determined by single-cell DNA-seq. After treatment with midostaurin for 3 months, significant reduction of KIT D816V and EZH2 mutations with significant expansion of TET2 and NRAS mutations was detected. The addition of azacitidine resulted in significant reduction of TET2 and NRAS mutations and progressive expansion of KIT D816V and EZH2 mutations. Acquisition of a new mutation (RUNX1 R162S) was followed by disease progression and rapid death after 8 months of combinatorial treatment. The results of single-cell DNA-seq are shown in a bar graph and fish plot with sequenced cell numbers listed below as combined blood and bone marrow cell count results. dRUNX1, double RUNX1 mutations.
Clonal evolution by serial bulk DNA next-generation sequencing and single-cell DNA-seq in a patient with advSM. (A) Clonal evolution in response to midostaurin and azacitidine by serial bulk DNA-seq. Founding ASXL1, SRSF2, and RUNX1 mutations persisted during midostaurin and azacitidine treatment. KIT and EZH2 mutation burden were initially high at the PB1 time point and were significantly diminished after midostaurin treatment. At the BM time point, allele frequency of NRAS and TET2 mutations were initially high, but they later decreased as a result of azacitidine treatment. KIT and EZH2 mutations, followed by disease relapse, recurred with an additional new mutation site at RUNX1 R162S. The results were projected into a trend line with VAF shown on the Y-axis and collected samples listed below the graph. (B) Clonal evolution in response to treatment with midostaurin and azacitidine determined by single-cell DNA-seq. After treatment with midostaurin for 3 months, significant reduction of KIT D816V and EZH2 mutations with significant expansion of TET2 and NRAS mutations was detected. The addition of azacitidine resulted in significant reduction of TET2 and NRAS mutations and progressive expansion of KIT D816V and EZH2 mutations. Acquisition of a new mutation (RUNX1 R162S) was followed by disease progression and rapid death after 8 months of combinatorial treatment. The results of single-cell DNA-seq are shown in a bar graph and fish plot with sequenced cell numbers listed below as combined blood and bone marrow cell count results. dRUNX1, double RUNX1 mutations.
In the single-cell DNA-seq data, we successfully detected all bulk-seq–verified mutations except the SRSF2 P95H and ASXL1 R693* mutations because there was no coverage by the DNA-seq amplicons at these 2 genomic loci (Table 2). All variants were annotated as acquired heterozygous mutations. In the BM sample, we identified a predominant and rapidly evolved KIT wild-type subclone with RUNX1 R204Q/TET2 P851Lfs*22/NRAS G12D co-mutations after treatment with midostaurin. This KIT wild-type subclone represented the CMML component in this SM patient that was dependent on NRAS signaling and was resistant to treatment with midostaurin. At the time of relapse, 2 major subclones were detected, each with RUNX1 R204Q/KIT D816V/EZH2 V696E/RUNX1 R162S and RUNX1 R204Q/TET2 P851Lfs*22 co-mutations. It is likely that these 2 subclones related to acquired resistance to midostaurin and azacitidine, respectively. The subclone with RUNX1 R204Q/TET2 P851Lfs*22/NRAS G12D co-mutations diminished significantly after treatment with azacitidine, indicating that the subclone that depended on NRAS signaling could be effectively controlled by azacitidine in this patient. By using single-cell DNA-seq data, a fish plot showing the clonal phylogeny was constructed to demonstrate both linear and branching clonal evolution patterns in advSM (Figure 1B).
Mutations detected by single-cell DNA sequencing in a patient with advSM
| Cell no. (subclone %) | |||
|---|---|---|---|
| Subclone variants | PB1 | BM (total N = 239)* | PB2 (total N = 4986)* |
| WT | NA | 2 (0.78) | 3626 (72.72) |
| RUNX1 R204Q | NA | 1 (0.39) | 63 (1.26) |
| RUNX1 R204Q/TET2 P851Lfs*22 | NA | 5 (1.95) | 513 (10.29) |
| RUNX1 R204Q/TET2 P851Lfs*22/NRAS G12D | NA | 231 (96.88) | 4 (0.08) |
| RUNX1 R204Q/KIT D816V | NA | 0 (0.00) | 33 (0.66) |
| RUNX1 R204Q/KIT D816V/EZH2 V696E | NA | 0 (0.00) | 181 (3.63) |
| RUNX1 R204Q/KIT D816V/EZH2 V696E/RUNX1 R162S | NA | 0 (0.00) | 566 (11.35) |
| Cell no. (subclone %) | |||
|---|---|---|---|
| Subclone variants | PB1 | BM (total N = 239)* | PB2 (total N = 4986)* |
| WT | NA | 2 (0.78) | 3626 (72.72) |
| RUNX1 R204Q | NA | 1 (0.39) | 63 (1.26) |
| RUNX1 R204Q/TET2 P851Lfs*22 | NA | 5 (1.95) | 513 (10.29) |
| RUNX1 R204Q/TET2 P851Lfs*22/NRAS G12D | NA | 231 (96.88) | 4 (0.08) |
| RUNX1 R204Q/KIT D816V | NA | 0 (0.00) | 33 (0.66) |
| RUNX1 R204Q/KIT D816V/EZH2 V696E | NA | 0 (0.00) | 181 (3.63) |
| RUNX1 R204Q/KIT D816V/EZH2 V696E/RUNX1 R162S | NA | 0 (0.00) | 566 (11.35) |
NA, not available.
*The number of cells with allele dropout or missing genotype in BM or PB2 was 331 and 1000, respectively.
To our knowledge, this was the first study to successfully unravel the clonal evolution and heterogeneity in response to selection pressure of KIT inhibition in advSM at single-cell resolution. Our results clearly show that progression in advSM can be caused by expansion of subclones acquiring new mutations in other myeloid genes independent of the KIT D816V mutation. In accordance with previous reports, no secondary KIT mutation related to drug resistance was detected.14 In addition, no copy number change in KIT or other myeloid genes was detected in the KIT D816V–mutated resistant subclone (supplemental Figure 1). Whether the activation of KIT D816V–independent signaling molecules such as Lyn and Btk or KIT D816V-dependent downstream pathways such as JAK/STAT and PI3K/AKT/mTOR is involved in the reemergence of a KIT-mutated subclone should be further explored in a future study.15 The rapid emergence of the KIT wild-type subclone after treatment with midostaurin also provides a molecular basis for the explanation of relative insensitivity of monocytes to KIT inhibitors in those with SM-CMML.13,16 Furthermore, the mutational history of driver genes as well as linear and branching clonal evolution were reconstructed in this patient with advSM. The detection of KIT wild-type and multimutated AHN subclones in advSM can be an important cause of acquired resistance to KIT inhibition and reinforces the concept that AHN-directed therapy will be necessary in addition to the KIT inhibitor. Besides, persistence of subclones with RUNX1, SRSF2, and ASXL1 mutations in early hematopoietic stem and progenitor cells probably linked to intrinsic resistant to midostaurin and azacitidine in advSM and importantly caused relapse.10 The use of other molecular targeted therapy such as SRSF2 inhibitor may be necessary to overcome primary resistant to the KIT inhibitor.17,18 In conclusion, our study has demonstrated the clonal evolution of malignant cells during the progression of ASM-CMML under treatment with midostaurin and azacitidine and provided a rationale for the combinatorial molecular targeted therapy in advSM.
To request data, please contact Ken-Hong Lim at limkenhong@gmail.com.
Acknowledgments:
The authors thank Renshiang Jhou (Prisma Biotech Corporation, Taipei, Taiwan) for help with the single-cell DNA sequencing and bioinformatics analysis.
This work was supported by a grant from the Ministry of Science and Technology, Taipei, Taiwan (MOST 107-2314-B-195-011-MY3) and intramural funding from the Department of Medical Research, MacKay Memorial Hospital.
Midostaurin was provided to the patient under a Novartis Managed Access Program.
Contribution: K.-H.L., J.-N.W., T.-Y.H., and Y.-C.C. conceived and designed the study; K.-H.L., C.G.-S.C., H.-C.L., and J.L. provided study materials or patient care; K.-H.L., J.-N.W., T.-Y.H., Y.-C.C., Y.-H.C., Y.-W.S., C.G.-S.C., and Y.-F.C. analyzed and interpreted the data; J.-Y.J. reviewed the bone marrow biopsies; K.-H.L. and J.-N.W. wrote the manuscript; and all authors provided final approval of the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Ken-Hong Lim, Division of Hematology and Oncology, MacKay Memorial Hospital, No. 92, Sec. 2, Zhongshan N Rd, Taipei City 104217, Taiwan; e-mail: limkenhong@gmail.com.

