

Key Points
Jmjd6-deficient HSCs progressively decline under steady-state conditions and fail to self-renew and regenerate hematopoiesis under stress.
JMJD6 suppresses pathways whose downregulation is fundamental for HSC integrity.

Abstract
Lifelong multilineage hematopoiesis critically depends on rare hematopoietic stem cells (HSCs) that reside in the hypoxic bone marrow microenvironment. Although the role of the canonical oxygen sensor hypoxia-inducible factor prolyl hydroxylase has been investigated extensively in hematopoiesis, the functional significance of other members of the 2-oxoglutarate (2-OG)-dependent protein hydroxylase family of enzymes remains poorly defined in HSC biology and multilineage hematopoiesis. Here, by using hematopoietic-specific conditional gene deletion, we reveal that the 2-OG–dependent protein hydroxylase JMJD6 is essential for short- and long-term maintenance of the HSC pool and multilineage hematopoiesis. Additionally, upon hematopoietic injury, Jmjd6-deficient HSCs display a striking failure to expand and regenerate the hematopoietic system. Moreover, HSCs lacking Jmjd6 lose multilineage reconstitution potential and self-renewal capacity upon serial transplantation. At the molecular level, we found that JMJD6 functions to repress multiple processes whose downregulation is essential for HSC integrity, including mitochondrial oxidative phosphorylation (OXPHOS), protein synthesis, p53 stabilization, cell cycle checkpoint progression, and mTORC1 signaling. Indeed, Jmjd6-deficient primitive hematopoietic cells display elevated basal and maximal mitochondrial respiration rates and increased reactive oxygen species (ROS), prerequisites for HSC failure. Notably, an antioxidant, N-acetyl-l-cysteine, rescued HSC and lymphoid progenitor cell depletion, indicating a causal impact of OXPHOS-mediated ROS generation upon Jmjd6 deletion. Thus, JMJD6 promotes HSC maintenance and multilineage differentiation potential by suppressing fundamental pathways whose activation is detrimental for HSC function.
Introduction
Multilineage hematopoiesis relies on hematopoietic stem cells (HSCs) residing within the hypoxic bone marrow (BM) microenvironment.1 Cellular responses to oxygen are predominantly mediated by hypoxia-inducible factor-α prolyl hydroxylase domain enzymes (PHDs), namely PHD1-3, which belong to the 2-oxoglutrate (2-OG)–dependent protein hydroxylase family.2 Indeed, conditional Phd2 deletion compromises HSC maintenance,3 and pharmacological PHD inhibition promotes HSC quiescence and enhances their mobilization from the BM.4,5 Notably, however, the functional significance of other 2-OG–dependent protein hydroxylases in HSCs and multilineage hematopoiesis remains elusive.
Here, we focus on JMJD6, a predominantly, though not exclusively, nuclear 2-OG–dependent protein hydroxylase with promiscuous substrate specificity.2,6,7 JMJD6-catalyzed protein hydroxylation is reported to be involved in the hypoxic response,8-10 RNA splicing,11,12 and regulation of gene transcription.13,14 JMJD6 has an essential function in embryonic development, with Jmjd6-null mice displaying perinatal lethality due to developmental defects of multiple organs, including kidney, intestine, liver, heart, and lungs.15,16 Furthermore, JMJD6 has been identified as a key regulator of tumorigenesis.7,17-19 However, the physiological significance of JMJD6 in normal adult tissue function remains poorly understood.
In our study, we used hematopoietic-specific conditional genetics to reveal JMJD6 as a fundamental regulator of adult HSC biology during unperturbed hematopoiesis, as well as upon hematopoietic injury and serial transplantation. Notably, we found that JMJD6 suppresses multiple pathways whose excessive activation is detrimental to normal hematopoiesis, including oxidative phosphorylation (OXPHOS)-mediated generation of reactive oxygen species (ROS).
Methods
Mice
All mice were on a C57BL/6 background, Vav-iCre mice20 have been described previously. The generation of Jmjd6fl mice is described in supplemental Figure 1. All transgenic and knockout mice were CD45.2+. Congenic recipient mice were CD45.1+/CD45.2+. All experiments involving mice were performed under University of Edinburgh and Queen Mary University of London Veterinary oversight with UK Home Office authorization. Treatment groups were randomized within boxes of littermates.
Flow cytometry
Peripheral blood (PB), BM, and splenic cells were prepared and analyzed as described previously.21-28 Hematopoietic stem and progenitor cell staining began with incubation with Fc block, followed by biotin-conjugated anti-lineage marker antibodies (anti-CD4, anti-CD5, anti-CD8a, anti-CD11b, anti-B220, anti–Gr-1, and anti-Ter119), allophycocyanin (APC)-conjugated anti–c-Kit, APC-Cy7–conjugated anti–Sca-1, phycoerythrin (PE)-conjugated anti-CD48, and PE-Cy7–conjugated anti-CD150 antibodies. Biotin-conjugated lineage markers were then stained with PerCP-conjugated streptavidin. For PB and differentiated cell analysis, cells were stained with PerCP-conjugated anti-B220, APC-Cy7–conjugated anti-CD19, APC-conjugated anti-CD11b, PE-Cy7–conjugated anti–Gr-1, PE-conjugated anti-CD8, and PE-conjugated anti-CD4 antibodies. To distinguish CD45.2 chimerism in transplantation experiments, fluorescein isothiocyanate–conjugated anti-CD45.1 and Pacific Blue–conjugated anti-CD45.2 antibodies were used. Flow cytometry analyses were performed using a LSRFortessa (BD). Cell sorting was performed on a FACSAria Fusion (BD).
Cell cycle analysis
BM samples were enriched for c-Kit+ cells and stained for surface markers, as described above, followed by fixation and permeabilization using Fixation/Permeabilization solution (BD Biosciences). The cells were then stained with PE-conjugated anti-Ki67 antibody (BioLegend). 4′,6-Diamidino-2-phenylindole (DAPI) was added before sample analysis.
Syngeneic transplantation assays
Transplant recipient CD45.1+/CD45.2+ mice were lethally irradiated with 2 5.5-Gy doses administered ≥4 hours apart, at an average rate of 0.58 Gy/min, using a 137Cs Gammacell 40 irradiator. For primary transplantations, lethally irradiated recipient CD45.1+/CD45.2+ mice were injected IV with 100 Lin−Sca-1+c-Kit+ (LSK)CD48−CD150+ HSCs and 200 000 support CD45.1+ unfractionated BM cells. For secondary transplantations, lethally irradiated recipient CD45.1+/CD45.2+ mice were injected IV with 4 × 106 unfractionated CD45.2+ BM cells from primary recipients and 200 000 support CD45.1+ unfractionated BM cells. All recipient mice were analyzed at 16 weeks posttransplantation.
CD117 (c-Kit) enrichment
Enrichment for CD117 (c-Kit)–expressing cells was performed using CD117 MicroBeads and LS columns (Miltenyi Biotec), according to the manufacturer’s instructions.
CFC assays
Colony-forming cell (CFC) assays were performed using MethoCult (M3434; STEMCELL Technologies). Two replicates were used per group in each experiment. For CFC assay experiments using N-acetyl-l-cysteine (NAC) (Sigma), NAC was dissolved in phosphate-buffered saline (PBS) and added at a concentration of 5 mM before plating.
Administration of 5-FU
Mice received 250 mg/kg of 5-fluorouracil (5-FU) via 1 IV injection. PBS was injected via 1 IV injection for control animals.
ROS analysis
BM cells were enriched for c-Kit+ cells and stained for ROS levels using CellROX reagent (Invitrogen), according to the manufacturer’s instructions.
Administration of NAC
Mice received 100 mg/kg NAC via daily intraperitoneal injection, as well as in drinking water (1 mg/mL) for the duration of the experiment. The water bottle containing NAC was changed twice per week.
Oxygen-consumption assays
Oxygen-consumption rates (OCRs) were measured using a Seahorse XF-24 analyzer (Seahorse Bioscience) and an XF Cell Mito Stress Test kit, as previously described.22 c-Kit+ cells from BM were plated in XF-24 microplates precoated with Cell-Tak (BD) at 250 000 cells per well in XF Base medium supplemented with 10 mM glucose, pH 7.4. OCR was measured 3 times every 6 minutes for basal value and after each sequential addition of oligomycin (1 µM), FCCP (1 µM), and rotenone and antimycin A (1 µM). Oxygen consumption measurements were normalized to cell counts before and after each assay.
Analysis of Jmjd6 expression in single-cell hematopoietic populations
To assess the differences in Jmjd6 expression among populations, we inspected 2 published single-cell data sets: a SMART-seq2 landscape of hematopoietic stem and progenitor populations29 and a 10× landscape of LSK and Lin−Sca-1−c-Kit+ (LK) populations.30 For both data sets, the corresponding bar plots of Jmjd6 expression were generated upon computing the average and the standard error of the mean (SEM) of the natural logarithm of the normalized expression of Jmjd6 in each population or cluster. All analyses were performed with the Scanpy python module.31
RNA-seq, GSEA, and differential splicing analysis
We assessed molecular consequences of Jmjd6 ablation in hematopoietic stem and progenitor cells from young Jmjd6CTL and Jmjd6cKO mice using RNA sequencing (RNA-seq). The use of LSK cells, as opposed to highly purified HSCs, allowed us to achieve a sufficient per-locus sequencing depth for robust global expression and splicing analyses, while minimizing the number of mice required. An average of 77.1 million 75-bp paired-end reads was sequenced per sample. Alignment to the GRCm38 mouse genome was performed with HISAT2 version 2.1.0,32 and read counts per gene were computed using the Rsubread package version 2.0.1 in R. Gene differential expression, identified using DESeq2 version 1.24, was ranked according to moderated t statistics, which take into account variability between genes in the ranking. Preranked genes were compared with gene lists in the Hallmark subset of the MSigDB database version 7.0 using gene set enrichment analysis (GSEA) software version 3.0 (http://software.broadinstitute.org/gsea) with 1000 permutations and default parameters. Splicing analysis was performed using DEXSeq version 1.30.0, limma version 3.40.6, and QoRTs-JunctionSeq version 1.14.0.
Results
JMJD6 is required for long-term HSC maintenance under steady-state conditions
To determine the expression of Jmjd6 in mouse stem and progenitor cells, we sorted LSK cells, LSKCD48−CD150+ HSCs, LSKCD48−CD150− multipotent progenitors (MPPs), primitive hematopoietic progenitor cells (HPCs; ie, LSKCD48+CD150− HPC-1 and LSKCD48+CD150+ HPC-2 populations), and LK myeloid progenitors and performed reverse transcription quantitative polymerase chain reaction. Jmjd6 was uniformly expressed among these populations (Figure 1A), with higher expression in MPPs and downregulation in LK myeloid progenitors. Additionally, to assess the expression of Jmjd6 in further hematopoietic compartments, we interrogated our SMART-seq2 single-cell expression data sets,29 and analysed Jmjd6 expression in long-term HSC (LSKCD34−CD135−), MPP1 (LSKCD34+CD135−CD150+CD48−), MPP2 (LSKCD34+CD135−CD150+CD48+), and MPP3 (LSKCD34+CD135−CD150−CD48+) populations, lymphoid-primed MPPs (LSKCD34+CD135+), which correspond to the MPP4 population,33 common myeloid progenitor (CMP; LKCD34+FcγRII/IIIlow), granulocyte-macrophage progenitor (GMP; LKCD34+FcγRII/IIIhigh), and megakaryocyte-erythroid progenitor (MEP; LKCD34−FcγRII/IIIlow) compartments. These analyses revealed that Jmjd6 was expressed rather uniformly across these populations (Figure 1B), with the highest expression in MEPs and the lowest expression in the MPP4 population. Finally, we used our 10× genomics single-cell RNA-seq30 to analyze Jmjd6 expression in HSCs and committed progenitor cell compartments, which revealed that Jmjd6 was expressed comparably among these populations (Figure 1C).
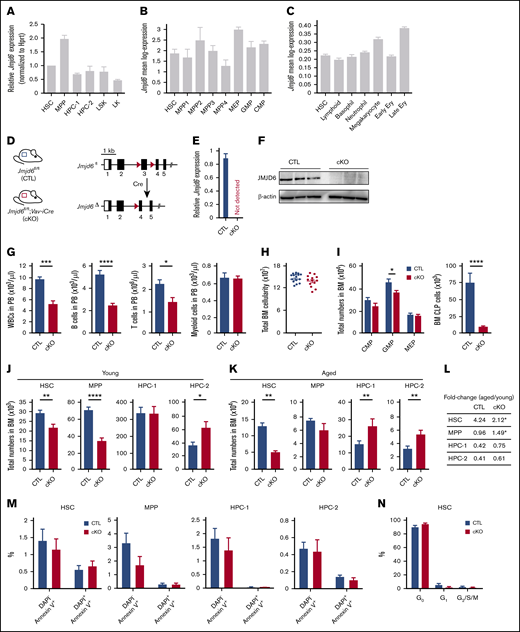
JMJD6 is required for steady-state multilineage hematopoiesis, whereas Jmjd6-deficient HSCs have a normal apoptotic and cell cycle status. (A) Relative levels of Jmjd6 messenger RNA (normalized to Hprt) in cells isolated from BM of 8- to 12-week-old adult C57BL/6 mice: LSKCD48−CD150+ HSCs, LSKCD48−CD150− MPPs, primitive HPCs (ie, LSKCD48+CD150− HPC-1 and LSKCD48+CD150+ HPC-2 populations), LSKs, and LK MPPs (n = 3). Data are mean ± SEM. (B) The expression of Jmjd6 in HSCs (LSKCD34−CD135−), MPP1 (LSKCD34+CD135−CD150+CD48−), MPP2 (LSKCD34+CD135−CD150+CD48+), MPP3 (LSKCD34+CD135−CD150−CD48+), and lymphoid-primed multipotent progenitor (LMPP)/MPP4 (LSKCD34+CD135+) populations, as well as in CMP (LKCD34+FcγRII/IIIlow), GMP (LKCD34+FcγRII/IIIhigh), and MEP (LKCD34−FcγRII/IIIlow) compartments, determined using single-cell SMART-seq2.29 Data are mean ± SEM. (C) Jmjd6 expression in HSCs and indicated committed progenitor cell compartments determined by 10× genomics single-cell RNA-seq.30 Bins represent clusters annotated via marker genes. Data are mean ± SEM. Ery, erythrocytes. (D) Genomic structure of the conditional Jmjd6 allele. Exon 3 is flanked by LoxP sites (red triangles). Following Cre-mediated recombination, exon 3 is excised, resulting in a frameshift mutation and a non-sense–mediated decay. (E) Levels of Jmjd6 messenger RNA in Jmjd6fl/fl;Vav-iCre (Jmjd6cKO) and control Jmjd6fl/fl (Jmjd6CTL) BM c-Kit+ cells (n = 4). Data are mean ± SEM. (F) Western blots for JMJD6 and β-actin from BM c-Kit+ cells from Jmjd6cKO and Jmjd6CTL mice (n = 3). (G) PB counts of white blood cells (WBCs), CD19+B220+ B cells, CD4+ and CD8+ T cells, and CD11b+Gr-1+ myeloid cells in 8- to 10-week-old Jmjd6cKO and Jmjd6CTL mice (n = 9-10). Data are mean ± SEM. (H) Total BM cellularity of Jmjd6cKO and Jmjd6CTL 8- to 10-week-old mice (2 femurs and 2 tibias) (n = 13-16). Data are mean ± SEM. (I) Total numbers of CMPs (LKCD34+FcγRII/IIIlow), GMPs (LKCD34+FcγRII/IIIhigh), MEPs (LKCD34−FcγRII/IIIlow), and CLPs (Lin−Sca-1lowc-KitlowCD127+CD135+) in BM from 8- to 10-week-old Jmjd6cKO and Jmjd6CTL mice (n = 13-16). Data are mean ± SEM. (J-K) Total numbers of HSCs, MPPs, HPC-1, and HPC-2 populations in BM of Jmjd6cKO and Jmjd6CTL mice. (J) Eight- to 10-week-old mice (n = 13-16). (K) Fifty-two–week-old mice (n = 6-9). Data are mean ± SEM. (L) Fold change in HSC, MPP, HPC-1, and HPC-2 populations in 52-week-old vs 8- to 10-week-old Jmjd6cKO and Jmjd6CTL mice (n = 6-13). (M) Percentage of DAPI−Annexin V+ and DAPI+Annexin V+ cells in HSCs, MPPs, HPC-1, and HPC-2 populations in BM from 8- to 10-week-old Jmjd6cKO and Jmjd6CTL mice (n = 6). Data are mean ± SEM. (N) Percentage of HSCs from 8- to 10-week-old Jmjd6cKO and Jmjd6CTL mice in the G0 (DAPI−Ki67−), G1 (DAPI−Ki67+), and G2/S/M (DAPI+Ki67+) phases of the cell cycle (n = 6-7). Data are mean ± SEM. *P < .05, **P < .01, ***P < .001, ****P < .0001, Mann-Whitney U test.
JMJD6 is required for steady-state multilineage hematopoiesis, whereas Jmjd6-deficient HSCs have a normal apoptotic and cell cycle status. (A) Relative levels of Jmjd6 messenger RNA (normalized to Hprt) in cells isolated from BM of 8- to 12-week-old adult C57BL/6 mice: LSKCD48−CD150+ HSCs, LSKCD48−CD150− MPPs, primitive HPCs (ie, LSKCD48+CD150− HPC-1 and LSKCD48+CD150+ HPC-2 populations), LSKs, and LK MPPs (n = 3). Data are mean ± SEM. (B) The expression of Jmjd6 in HSCs (LSKCD34−CD135−), MPP1 (LSKCD34+CD135−CD150+CD48−), MPP2 (LSKCD34+CD135−CD150+CD48+), MPP3 (LSKCD34+CD135−CD150−CD48+), and lymphoid-primed multipotent progenitor (LMPP)/MPP4 (LSKCD34+CD135+) populations, as well as in CMP (LKCD34+FcγRII/IIIlow), GMP (LKCD34+FcγRII/IIIhigh), and MEP (LKCD34−FcγRII/IIIlow) compartments, determined using single-cell SMART-seq2.29 Data are mean ± SEM. (C) Jmjd6 expression in HSCs and indicated committed progenitor cell compartments determined by 10× genomics single-cell RNA-seq.30 Bins represent clusters annotated via marker genes. Data are mean ± SEM. Ery, erythrocytes. (D) Genomic structure of the conditional Jmjd6 allele. Exon 3 is flanked by LoxP sites (red triangles). Following Cre-mediated recombination, exon 3 is excised, resulting in a frameshift mutation and a non-sense–mediated decay. (E) Levels of Jmjd6 messenger RNA in Jmjd6fl/fl;Vav-iCre (Jmjd6cKO) and control Jmjd6fl/fl (Jmjd6CTL) BM c-Kit+ cells (n = 4). Data are mean ± SEM. (F) Western blots for JMJD6 and β-actin from BM c-Kit+ cells from Jmjd6cKO and Jmjd6CTL mice (n = 3). (G) PB counts of white blood cells (WBCs), CD19+B220+ B cells, CD4+ and CD8+ T cells, and CD11b+Gr-1+ myeloid cells in 8- to 10-week-old Jmjd6cKO and Jmjd6CTL mice (n = 9-10). Data are mean ± SEM. (H) Total BM cellularity of Jmjd6cKO and Jmjd6CTL 8- to 10-week-old mice (2 femurs and 2 tibias) (n = 13-16). Data are mean ± SEM. (I) Total numbers of CMPs (LKCD34+FcγRII/IIIlow), GMPs (LKCD34+FcγRII/IIIhigh), MEPs (LKCD34−FcγRII/IIIlow), and CLPs (Lin−Sca-1lowc-KitlowCD127+CD135+) in BM from 8- to 10-week-old Jmjd6cKO and Jmjd6CTL mice (n = 13-16). Data are mean ± SEM. (J-K) Total numbers of HSCs, MPPs, HPC-1, and HPC-2 populations in BM of Jmjd6cKO and Jmjd6CTL mice. (J) Eight- to 10-week-old mice (n = 13-16). (K) Fifty-two–week-old mice (n = 6-9). Data are mean ± SEM. (L) Fold change in HSC, MPP, HPC-1, and HPC-2 populations in 52-week-old vs 8- to 10-week-old Jmjd6cKO and Jmjd6CTL mice (n = 6-13). (M) Percentage of DAPI−Annexin V+ and DAPI+Annexin V+ cells in HSCs, MPPs, HPC-1, and HPC-2 populations in BM from 8- to 10-week-old Jmjd6cKO and Jmjd6CTL mice (n = 6). Data are mean ± SEM. (N) Percentage of HSCs from 8- to 10-week-old Jmjd6cKO and Jmjd6CTL mice in the G0 (DAPI−Ki67−), G1 (DAPI−Ki67+), and G2/S/M (DAPI+Ki67+) phases of the cell cycle (n = 6-7). Data are mean ± SEM. *P < .05, **P < .01, ***P < .001, ****P < .0001, Mann-Whitney U test.
To determine the requirement for Jmjd6 in HSC maintenance and multilineage hematopoiesis, we generated a floxed Jmjd6 allele in which exon 3 (encoding the catalytic domain) is flanked by LoxP sites (supplemental Figure 1). We combined the Jmjd6fl allele with Vav-iCre20 (Figure 1D; supplemental Figure 1) to generate Jmjd6fl/fl;Vav-iCre (Jmjd6cKO) mice, in which Jmjd6 is specifically deleted within the hematopoietic system. The absence of Jmjd6 from the hematopoietic system was confirmed at the transcript and protein levels (Figure 1E-F). Notably, Jmjd6 deletion had no impact on murine viability, with Jmjd6cKO and control Jmjd6fl/fl (Jmjd6CTL) mice born at normal Mendelian ratios, allowing us to investigate the functional significance of Jmjd6 in adult HSC biology and multilineage hematopoiesis.
PB analyses of Jmjd6cKO mice revealed a drastic reduction in the numbers of white blood cells, B cells, and T cells, whereas the numbers of erythrocytes and myeloid cells were unaffected (Figure 1G; supplemental Figure 2A). BM and spleens from Jmjd6cKO mice exhibited normal total cellularity and largely unaffected differentiated cell counts (Figures 1H; supplemental Figure 2B-C). Notably, BM GMPs and common lymphoid progenitors (CLPs) were markedly decreased, whereas CMPs and MEPs were unchanged (Figure 1I). Strikingly, although Jmjd6-deficient BM cells displayed normal differentiation in CFC assays, they failed to form secondary colonies after replating (supplemental Figure 2D). Thus, loss of JMJD6 has a minimal impact on myeloid differentiation, but depletes CLPs and compromises lymphoid lineage output under steady-state conditions.
Having discovered a decrease in GMPs and CLPs upon Jmjd6 deletion, as well as failure of colony formation upon replating, we next investigated the impact of Jmjd6 deficiency at the top of the hematopoietic differentiation hierarchy. Eight- to 12-week-old Jmjd6cKO mice displayed a significant depletion of HSC and MPP cell pools compared with Jmjd6CTL mice (Figure 1J; supplemental Figure 2E). Furthermore, to test the impact of Jmjd6 deletion on long-term HSC maintenance under steady-state conditions, we aged Jmjd6cKO and Jmjd6CTL mice for 52 weeks and found that aged Jmjd6cKO mice exhibited a more pronounced HSC loss than did young Jmjd6cKO mice, relative to age-matched controls (Figure 1K-L; supplemental Figure 3). Consistent with an increase in HSC numbers upon physiological aging,34 we found that the HSC pool in aged Jmjd6CTL mice expanded 4.24-fold vs young animals, whereas the HSC pool in Jmjd6cKO mice expanded only 2.12-fold (Figure 1L). Notably, despite a marked decrease in HSC numbers, aged Jmjd6cKO mice did not display any other hematopoietic defects (supplemental Figure 3B-D), suggesting the activation of compensatory mechanisms upon aging to bypass phenotypes (including a decrease in CLPs and GMPs, and lymphoid defects) observed in young Jmjd6cKO mice. Taken together, JMJD6 is essential for long-term cell-autonomous maintenance of the HSC pool during unperturbed hematopoiesis.
HSCs lacking Jmjd6 have normal survival rate and remain quiescent
Given that HSC depletion may result from an increase in cell death or a loss of HSC quiescence,35,36 we investigated whether the reduction in HSCs upon Jmjd6 deletion is associated with increased apoptosis or decreased quiescence. To determine the rate of cell death, we analyzed primitive hematopoietic compartments using DAPI and Annexin V. The percentages of DAPI−Annexin V+ (early apoptotic) or DAPI+Annexin V+ (late apoptotic) cells were comparable between Jmjd6CTL and Jmjd6cKO mice (Figure 1M). Furthermore, to determine the cell cycle status of Jmjd6-deficient HSCs, we used Ki-67 and DAPI staining, which did not reveal any differences in the quiescence of Jmjd6CTL and Jmjd6cKO HSCs (Figure 1N). Therefore, HSC depletion upon Jmjd6 loss is unlikely to result from increased HSC apoptosis or their loss of quiescence.
Jmjd6 deletion delays hematopoietic cell recovery following myeloablation
Given the progressive loss of Jmjd6-deficient HSCs, we next investigated their regenerative capacity upon hematopoietic injury. Young 8- to 12-week-old Jmjd6CTL and Jmjd6cKO mice were administered a single dose of 5-FU and analyzed 10 and 14 days postinjection (Figure 2A). At day 10 following 5-FU treatment, Jmjd6cKO mice displayed a marked decrease in total BM cellularity and BM differentiated cells (Figure 2B-C), as well as near-complete ablation of myeloid progenitors (Figure 2D), HSCs, and primitive progenitor cell compartments (Figure 2E-G) compared with Jmjd6CTL mice. Analyses at day 14 posttreatment revealed a decrease in differentiated cell numbers in Jmjd6cKO mice (Figure 2B-C) vs Jmjd6CTL mice; however, the difference was notably less marked than at day 10 posttreatment. Strikingly, at day 14 posttreatment, we observed a robust recovery of the LK, LSK, HSC, HPC-1, and HPC-2 cell compartments in Jmjd6cKO mice, whereas the recovery of MPP cells remained defective (Figure 2D-G). Overall, based on these data, we conclude that JMJD6 deficiency delays the recovery of hematopoietic cells at the primitive and differentiated hierarchical levels following myeloablation.
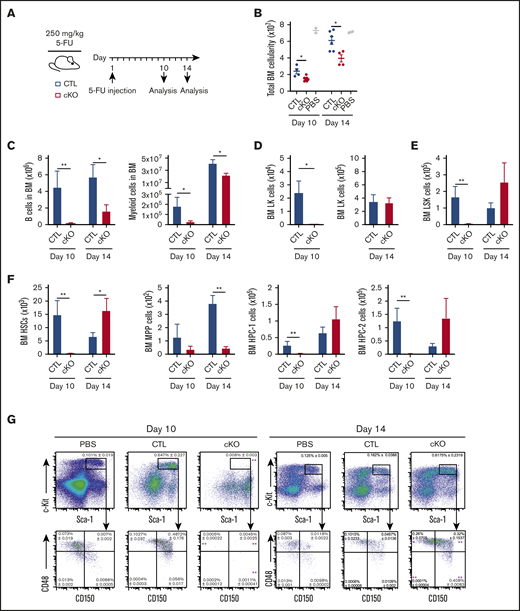
Loss of Jmjd6 significantly impacts recovery from hematopoietic injury. (A) Experimental design. Eight- to 12-week-old Jmjd6cKO and Jmjd6CTL mice were administered 1 dose of 5-FU (250 mg/kg) via IV injection, and hematopoietic compartments were analyzed 10 and 14 days later. (B) Total BM cellularity (1 femur and 1 tibia) of 5-FU–treated Jmjd6cKO and Jmjd6CTL mice (n = 4-7) and Jmjd6CTL PBS-treated controls (n = 2) at 10 and 14 days postinjection. Total cell numbers of B cells and myeloid cells (C), LK cells (D), LSK cells (E), and HSCs, MPPs, and HPC-1 and HPC-2 populations (F) in BM from Jmjd6cKO and Jmjd6CTL mice 10 and 14 days following 5-FU treatment (n = 4-7). (G) Representative FACS profiles showing frequencies (± SEM) of BM LSK, HSC, MPP, HPC-1, and HPC-2 cell populations from 5-FU–treated Jmjd6cKO and Jmjd6CTL mice (n = 4-7) and Jmjd6CTL PBS-treated controls (n = 2) at 10 and 14 days postinjection. Data are mean ± SEM. *P < .05, **P < .01, Mann-Whitney U test.
Loss of Jmjd6 significantly impacts recovery from hematopoietic injury. (A) Experimental design. Eight- to 12-week-old Jmjd6cKO and Jmjd6CTL mice were administered 1 dose of 5-FU (250 mg/kg) via IV injection, and hematopoietic compartments were analyzed 10 and 14 days later. (B) Total BM cellularity (1 femur and 1 tibia) of 5-FU–treated Jmjd6cKO and Jmjd6CTL mice (n = 4-7) and Jmjd6CTL PBS-treated controls (n = 2) at 10 and 14 days postinjection. Total cell numbers of B cells and myeloid cells (C), LK cells (D), LSK cells (E), and HSCs, MPPs, and HPC-1 and HPC-2 populations (F) in BM from Jmjd6cKO and Jmjd6CTL mice 10 and 14 days following 5-FU treatment (n = 4-7). (G) Representative FACS profiles showing frequencies (± SEM) of BM LSK, HSC, MPP, HPC-1, and HPC-2 cell populations from 5-FU–treated Jmjd6cKO and Jmjd6CTL mice (n = 4-7) and Jmjd6CTL PBS-treated controls (n = 2) at 10 and 14 days postinjection. Data are mean ± SEM. *P < .05, **P < .01, Mann-Whitney U test.
JMJD6 is essential for HSC maintenance upon serial transplantation
Considering the significant HSC depletion in steady-state conditions, as well as the delay in the regeneration of Jmjd6-deficient HSCs upon hematopoietic injury, we next tested the multilineage reconstitution capacity of Jmjd6cKO HSCs upon serial transplantation (Figure 3A). First, we competitively transplanted HSCs from young Jmjd6cKO and Jmjd6CTL mice into lethally irradiated primary recipients and found that Jmjd6-deficient HSCs displayed reduced donor-derived chimerism compared with control HSCs (Figure 3B). Moreover, Jmjd6cKO HSCs retained their myeloid-reconstitution capacity but lost their lymphoid lineage–differentiation potential, suggesting a myeloid bias (Figure 3B). Notably, although Jmjd6cKO HSC and MPP populations were reduced under steady-state conditions (Figure 1J), Jmjd6cKO HSCs contributed to the HSC and primitive progenitor cell compartments of primary recipients in a comparable manner to Jmjd6CTL HSCs (Figure 3C). Strikingly, however, secondary transplantation unveiled a dramatic failure of Jmjd6-deficient HSCs to repopulate short- and long-term multilineage hematopoiesis, with Jmjd6cKO HSCs unable to contribute to the PB compartment, and a significant reduction in the contribution to BM primitive and progenitor compartments compared with Jmjd6CTL HSCs (Figure 3D-E). Therefore, JMJD6 is an essential regulator of HSC self-renewal and posttransplantation multilineage hematopoiesis.
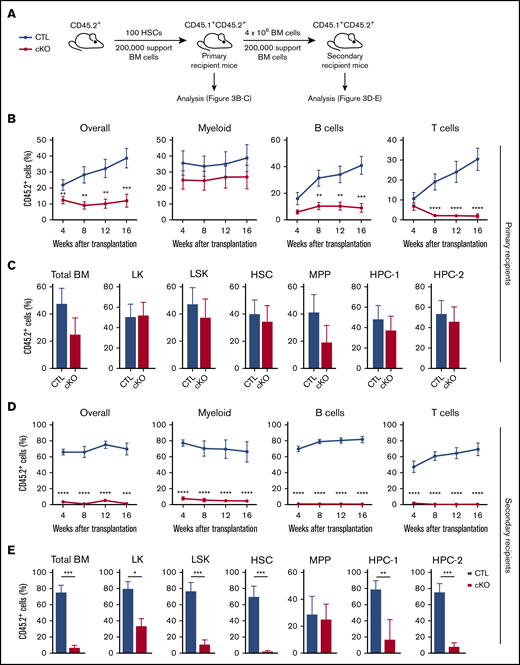
HSCs without JMJD6 fail to sustain multilineage hematopoiesis upon serial transplantation. (A) Experimental design. One hundred BM HSCs from 8- to 10-week-old Jmjd6cKO and Jmjd6CTL mice were transplanted into lethally irradiated syngeneic CD45.1+/CD45.2+ primary recipient mice together with 2 × 105 competitor CD45.1+ BM cells. After 16 weeks, 4 × 106 unfractionated BM cells from primary recipient mice were transplanted into lethally irradiated syngeneic CD45.1+/CD45.2+ secondary recipient mice together with 2 × 105 competitor CD45.1+ BM cells. PB of primary and secondary recipient mice was analyzed every 4 weeks and hematopoietic compartments were analyzed 16 weeks posttransplantation. (B) Percentage of CD45.2+ cells in the overall PB compartment, as well as the myeloid, B-cell, and T-cell compartments, in primary recipient mice (n = 2 per genotype; 4-5 recipients per group). (C) Percentage of CD45.2+ cells within the total BM, LK, LSK, HSC, MPP, HPC-1, and HPC-2 compartments of primary recipient mice 16 weeks after transplantation (n = 2 per genotype; 4-5 recipients per group). (D) Percentage of CD45.2+ cells in the overall PB compartment, as well as the myeloid, B-cell, and T-cell compartments, in secondary recipient mice (n = 2 per genotype; 4-5 recipients per group). (E) Percentage of CD45.2+ cells within the total BM, LK, LSK, HSC, MPP, HPC-1, and HPC-2 compartments of secondary recipient mice 16 weeks after transplantation (n = 2 per genotype; 4-5 recipients per group). Data represent mean ± SEM. *P < .05, **P < .01, ***P < .001, ****P < .0001, Mann-Whitney U test.
HSCs without JMJD6 fail to sustain multilineage hematopoiesis upon serial transplantation. (A) Experimental design. One hundred BM HSCs from 8- to 10-week-old Jmjd6cKO and Jmjd6CTL mice were transplanted into lethally irradiated syngeneic CD45.1+/CD45.2+ primary recipient mice together with 2 × 105 competitor CD45.1+ BM cells. After 16 weeks, 4 × 106 unfractionated BM cells from primary recipient mice were transplanted into lethally irradiated syngeneic CD45.1+/CD45.2+ secondary recipient mice together with 2 × 105 competitor CD45.1+ BM cells. PB of primary and secondary recipient mice was analyzed every 4 weeks and hematopoietic compartments were analyzed 16 weeks posttransplantation. (B) Percentage of CD45.2+ cells in the overall PB compartment, as well as the myeloid, B-cell, and T-cell compartments, in primary recipient mice (n = 2 per genotype; 4-5 recipients per group). (C) Percentage of CD45.2+ cells within the total BM, LK, LSK, HSC, MPP, HPC-1, and HPC-2 compartments of primary recipient mice 16 weeks after transplantation (n = 2 per genotype; 4-5 recipients per group). (D) Percentage of CD45.2+ cells in the overall PB compartment, as well as the myeloid, B-cell, and T-cell compartments, in secondary recipient mice (n = 2 per genotype; 4-5 recipients per group). (E) Percentage of CD45.2+ cells within the total BM, LK, LSK, HSC, MPP, HPC-1, and HPC-2 compartments of secondary recipient mice 16 weeks after transplantation (n = 2 per genotype; 4-5 recipients per group). Data represent mean ± SEM. *P < .05, **P < .01, ***P < .001, ****P < .0001, Mann-Whitney U test.
JMJD6 suppresses OXPHOS and ROS generation to control hematopoiesis
To mechanistically understand the failure of Jmjd6-deficient HSCs upon serial transplantation, we examined the molecular signature of Jmjd6-deficient LSK cells. Although differential gene expression analysis identified a number of individual deregulated genes (supplemental Figure 4), GSEA indicated a broader upregulation of processes detrimental to HSCs, including p53 activity35,37 (which is known to be directly repressed by JMJD67,17 ), G1/S and G2/M checkpoints, OXPHOS,38 mTORC1 signaling,39 protein synthesis,40 and E2F signaling41 (Figure 4A-B). Given that suppression of OXPHOS is essential for HSC self-renewal, and its upregulation devastates HSC functions,38 we validated the impact of Jmjd6 deletion on OXPHOS using a Seahorse XF Analyzer. We found that the basal and maximal OCRs were significantly increased in Jmjd6-deficient cells (Figure 4C). Thus, Jmjd6 deletion results in upregulation of multiple pathways, including activation of OXPHOS, whose excessive activation is known to have detrimental consequences to HSCs.
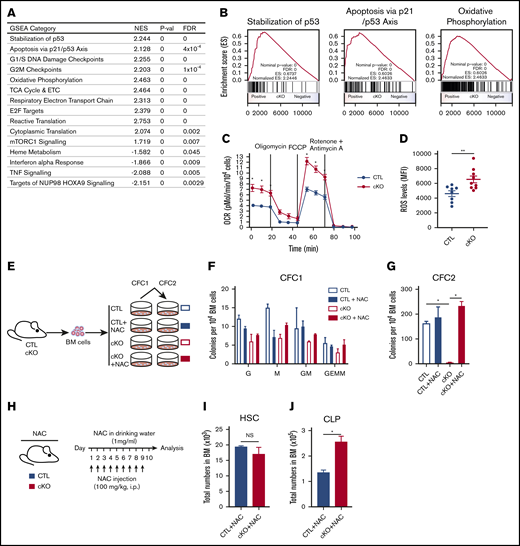
Jmjd6-deficient HSPCs display molecular signatures of functional HSC decline, which can be rescued by removing ROS. (A) GSEA showing upregulated and downregulated pathways in LSKs from 8- to 12-week-old Jmjd6cKO mice compared with LSKs from 8- to 12-week-old Jmjd6CTL mice (n = 4). (B) GSEA plots for stabilization of p53, apoptosis via p21/p53 axis, and OXPHOS based on analysis of gene expression changes, using Jmjd6cKO LSK cells for upregulated pathways (n = 4). (C) OCR in Jmjd6cKO and Jmjd6CTL BM c-Kit+ cells under basal conditions and maximal OCR. Maximal OCR was achieved by the sequential addition of oligomycin (ATPase inhibitor), FCCP (mitochondrial uncoupler), and rotenone and antimycin A (complex I and III inhibitors, respectively) (n = 3). (D) ROS levels in c-Kit+Jmjd6cKO and Jmjd6CTL BM cells after a 1-hour incubation with CellROX reagent. Data represent mean fluorescence intensity (MFI) (n = 8-9). (E) Experimental design. BM cells from 8- to 10-week-old Jmjd6cKO and Jmjd6CTL mice were placed in CFC assays with 5 mM NAC or PBS control. CFC1 colonies were counted and scored 10 days after plating and replated into CFC2. CFC2 colonies were counted after 10 days in culture. (F) CFC1. G, granulocyte; GEMM, granulocyte, erythroid, macrophage, or megakaryocyte; GM, granulocyte and monocyte/macrophage; M, monocyte/macrophage; (n = 4). (G) CFC 2 (n = 4). (H) Experimental design. Eight- to 12-week-old Jmjd6cKO and Jmjd6CTL mice were administered a daily dose of NAC (100 mg/kg) via intraperitoneal injection, as well as in drinking water (1 mg/mL). Hematopoietic compartments were analyzed following 10 days of treatment. Total cell numbers of HSCs (I) and CLPs (J) in BM from Jmjd6cKO and Jmjd6CTL mice after 10 days of NAC treatment (n = 4-5). Data are mean ± SEM. *P < .05, **P < .01. NS, not significant.
Jmjd6-deficient HSPCs display molecular signatures of functional HSC decline, which can be rescued by removing ROS. (A) GSEA showing upregulated and downregulated pathways in LSKs from 8- to 12-week-old Jmjd6cKO mice compared with LSKs from 8- to 12-week-old Jmjd6CTL mice (n = 4). (B) GSEA plots for stabilization of p53, apoptosis via p21/p53 axis, and OXPHOS based on analysis of gene expression changes, using Jmjd6cKO LSK cells for upregulated pathways (n = 4). (C) OCR in Jmjd6cKO and Jmjd6CTL BM c-Kit+ cells under basal conditions and maximal OCR. Maximal OCR was achieved by the sequential addition of oligomycin (ATPase inhibitor), FCCP (mitochondrial uncoupler), and rotenone and antimycin A (complex I and III inhibitors, respectively) (n = 3). (D) ROS levels in c-Kit+Jmjd6cKO and Jmjd6CTL BM cells after a 1-hour incubation with CellROX reagent. Data represent mean fluorescence intensity (MFI) (n = 8-9). (E) Experimental design. BM cells from 8- to 10-week-old Jmjd6cKO and Jmjd6CTL mice were placed in CFC assays with 5 mM NAC or PBS control. CFC1 colonies were counted and scored 10 days after plating and replated into CFC2. CFC2 colonies were counted after 10 days in culture. (F) CFC1. G, granulocyte; GEMM, granulocyte, erythroid, macrophage, or megakaryocyte; GM, granulocyte and monocyte/macrophage; M, monocyte/macrophage; (n = 4). (G) CFC 2 (n = 4). (H) Experimental design. Eight- to 12-week-old Jmjd6cKO and Jmjd6CTL mice were administered a daily dose of NAC (100 mg/kg) via intraperitoneal injection, as well as in drinking water (1 mg/mL). Hematopoietic compartments were analyzed following 10 days of treatment. Total cell numbers of HSCs (I) and CLPs (J) in BM from Jmjd6cKO and Jmjd6CTL mice after 10 days of NAC treatment (n = 4-5). Data are mean ± SEM. *P < .05, **P < .01. NS, not significant.
Given that elevated mitochondrial respiration frequently contributes to the formation of ROS,38,42 we investigated whether enhanced OXPHOS in Jmjd6-deficient cells results in increased ROS generation. Using CellROX to detect and quantify ROS levels, we discovered that c-Kit+ cells from Jmjd6cKO mice had significantly increased levels of ROS compared with control cells (Figure 4D). Given that elevated ROS have detrimental consequences to hematopoietic cells,42,43 we next tested whether the antioxidant NAC could alleviate the hematopoietic phenotypes resulting from Jmjd6 deficiency. To investigate this, we serially plated BM cells from Jmjd6CTL and Jmjd6cKO mice into CFC assays, in the presence or absence of NAC (Figure 4E). As expected, Jmjd6CTL and Jmjd6cKO cells efficiently generated primary colonies in control and NAC-containing methylcellulose cultures (Figure 4F). However, although Jmjd6-deficient cells failed to replate in the absence of NAC, strikingly, Jmjd6-deficient cells incubated with NAC efficiently produced colony numbers that were comparable to control cells (Figure 4G). Finally, to determine the significance of elevated ROS upon Jmjd6 deletion in vivo, we treated Jmjd6CTL and Jmjd6cKO mice with NAC for 10 days (Figure 4H). Significantly, we discovered that Jmjd6cKO mice treated with NAC no longer displayed reduced numbers of HSC and CLP cell populations (Figure 4I-J). Taken together, we conclude that elevated ROS in Jmjd6-deficient cells is causal for their failure to generate hematopoietic colonies upon replating, as well as contributing to reduced HSC and CLP numbers in Jmjd6cKO mice in vivo.
JMJD6 does not regulate messenger RNA splicing in HSPCs
Finally, JMJD6 is known to catalyze hydroxylation of splicing regulators, including U2A65 (2AF2), and has been shown to regulate splicing in vivo in developing tissues and the thymus.44,45 We therefore asked to what extent Jmjd6 deletion impacts alternative splicing. In spite of substantial RNA-seq coverage, only 7 transcripts showed an effect in LSK cells (supplemental \ure 5). This result indicates that JMJD6 is unlikely to function as a major regulator of alternative splicing in HSCs and progenitors.
Discussion
Despite extensive biochemical studies, the functional significance of JMJD6 in normal adult tissues remains poorly understood. JMJD6 is a predominantly nuclear protein, which is reported to regulate transcription and messenger RNA splicing.11,13,14 At the molecular level, JMJD6 catalyzes hydroxylation of lysine residues of multiple substrates, including p53,7,17 pVHL,7,9,10 ERα,13 and splicing regulators, including U2AF65.12,44 Our analyses revealed that loss of JMJD6 results in HSC depletion and functional failure of HSCs to sustain self-renewal and hematopoietic regeneration. Furthermore, loss of Jmjd6 exposes HSCs to molecular vulnerabilities, upregulating multiple pathways whose suppression is essential for normal HSC functions, including OXPHOS.38 Consistent with increased mitochondrial OXPHOS upon Jmjd6 deletion, which predisposes to ROS generation,38,42 ROS were causal, at least in part, for HSC and CLP depletion resulting from Jmjd6 loss, implying that, under physiological conditions, JMJD6 may function to repress OXPHOS-generated ROS to control hematopoiesis. Furthermore, consistent with direct suppression of p53 by JMJD6,7,17 we found that Jmjd6-deficient cells upregulated p53-related signatures, whose activation promotes HSC loss.35,37 We also found that Jmjd6 deficiency results in upregulation of other pathways whose activity is known to deplete or exhaust HSCs, including mTORC1 signaling,39 protein synthesis,40 and E2F downstream pathway.41 Notably, although our findings reveal JMJD6 as a suppressor of multiple pathways, whose excessive activation promotes HSC failure, the mechanisms through which JMJD6 functions in this context remain an open question, meriting further investigations. Finally, we revealed that JMJD6 is unlikely to be a major splicing regulator in HSCs. Rather, JMJD6 functions to promote the long-term maintenance of HSCs, their regenerative capacity, and self-renewal potential by restraining multiple pathways, including OXPHOS-mediated ROS generation, whose strict control is essential for their integrity.
Data sharing requests should be sent to Kamil R. Kranc (kamil.kranc@qmul.ac.uk).
Acknowledgments
The authors thank Fiona K. Hamey for establishing a pipeline for single-cell expression analyses and are extremely grateful to all members of the Biological Services Unit at Queen Mary University of London for exemplary dedication to this research during the COVID-19 pandemic. The authors thank Vladimir Benes and Jelena Pistolic (Genomics Core facility, European Molecular Biology Laboratory, Heidelberg, Germany) for performing the gene expression profiling.
This work was supported by a project grant from Blood Cancer UK (formerly Bloodwise). K.R.K.’s laboratory is also supported by a Cancer Research UK Programme Grant, The Barts Charity, the Medical Research Council, and the Kay Kendall Leukaemia Fund. N.M.M. was funded by a Wellcome Trust New Investigator Award. A.L. and D.V. received support from the Biotechnology and Biological Sciences Research Council Institute Strategic Program Funding. D.V.'s laboratory was also supported by Kay Kendall Leukaemia Fund.
Authorship
Contribution: K.R.K. funded and designed the experiments and wrote the manuscript; H.L. performed in vivo and in vitro experiments, FACS and data analyses, and wrote the manuscript; C.S. and J.D. performed in vivo and in vitro experiments, FACS, and data analyses; L.N.v.d.L. performed computational analyses of gene expression and splicing; M.B. performed single-cell expression analyses; A.T., E.G., A.S., P.T., R.N.C., L.A., J.C., M.V., A.V.G., and P.G. helped with in vivo and in vitro experiments, FACS, and data analyses; D.V., N.M.M., N.P.R., B.G., C.J.S., and D.O. provided significant scientific expertise to this study; and A.L. and M.O. produced Jmjd6fl mice.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Kamil R. Kranc, Laboratory of Haematopoietic Stem Cell & Leukaemia Biology, Centre for Haemato-Oncology, Barts Cancer Institute, Queen Mary University of London, Charterhouse Square, London EC1M 6BQ, United Kingdom; e-mail: kamil.kranc@qmul.ac.uk.

