

Key Points
Rh antibodies are the most common antibodies in patients with transfusion-dependent thalassemia despite RhD, C, E, and K matched transfusions.
Rh alloimmunization that is not explained by the patients’ RH genotypes suggest a role of variant Rh antigens on Black donor red cells.

Abstract
Chronically transfused patients with thalassemia are at risk for red cell alloimmunization. No studies have specifically examined alloimmunization after implementation of prophylactic Rh (D, C, E) and K matched red cells in a racially diverse population of thalassemia patients and donors. This retrospective study examined Rh antibodies among 40 chronically transfused patients (Asian, White, Black, Indian, Middle Eastern) with thalassemia receiving a mean of 174 serologic prophylactic RhD, C, E, and K matched red cell units. We examined the patients’ RH genotype, as well as donor race and Rh phenotypes over 3 transfusion events preceding antibody detection. Eighteen alloantibodies were detected in 13 of 40 patients (32.5%), with an alloimmunization rate of 0.26 antibodies per 100 units transfused. Thirteen antibodies (72.2%) were directed against Rh (5 anti-D, 4 anti-C, 2 anti-E, 1 anti-e, 1 anti-V), despite donor phenotypes that confirmed lack of transfusion of D, C, or E antigens to patients lacking the corresponding antigen(s). Ten of 40 patients had an altered RH genotype, but the Rh antibodies were not associated with patients with variant RH. Black donors with a known high frequency of RH variants provided 63% of the units transfused in the 3 visits preceding unexplained anti-Rh detection. Rh alloimmunization not explained by the thalassemia patients’ RH genotype or the donors’ serologic phenotype suggests more precise matching is needed, and the role of donor RH genotypes on alloimmunization should be explored. Extending Rh D, C, and E matching to include c and e would result in better-matched units and further minimize Rh alloimmunization.
Introduction
Chronic life-long transfusions are often required for patients with severe thalassemia to provide normal red blood cells and to suppress the patients’ own ineffective erythropoiesis. Cumulative exposure increases the risk for red cell alloimmunization and subsequent delayed hemolytic transfusion reactions (DHTRs).1 The prevalence of alloimmunization among patients with thalassemia ranges from 3% to 42%, with most antibodies directed against the Rh system.2 In the United States, varying antigen-matching practices contribute to the wide range of alloimmunization prevalence reported and includes ABO RhD–matched, prophylactic ABO RhD, C, E and K matched (CEK), CcEe and K matched, and/or additional extended matched to include JK, FY, and S/s.3-5 Some institutions provide prophylactic Rh and K or extended matched red cells only after an individual has become alloimmunized. An international consensus group recommends prophylactic ABO, RhD, Cc, Ee, and K matched red cells for individuals with thalassemia, even in the absence of alloantibodies, to reduce the risk of alloimmunization.6 However, the recommendation was based on very-low-quality studies, and therefore, the strength of the recommendation was weak according to the Grading of Recommendations, Assessment, Development and Evaluation tool used.7 To date, no studies have specifically examined alloimmunization in individuals with thalassemia of diverse racial backgrounds receiving prophylactic RhD, C, E, and K matched red cell transfusions.2
The Rh blood group system is not only the most immunogenic following ABO, but it is highly complex with >50 serologically defined antigens and >500 RHD and RHCE alleles identified. Among European-based populations, the frequency of variant RH alleles is ∼1% to 2%, whereas more than 85% of individuals of African Black ancestry have at least 1 RH variant.8,9 RH variation among Asians is uncommon, and the overall population frequency has not been comprehensively documented.10 Variant alleles may result in partial Rh antigen expression in which some epitopes are lacking and may lead an individual to recognize the conventional antigen as foreign.11 Standard serologic red cell typing does not distinguish variation in expression of the common Rh antigens (D, C, c, E, e),8 such that serologic Rh matching reduces but does not eliminate Rh alloantibody formation in patients with sickle cell disease (SCD).9,12 Despite prophylactic Rh (D, C, E) and K matching for the past 25 years at our institution, Rh antibodies continue to be the most common specificities identified in chronically transfused patients with thalassemia. The specific impact of RH variation in patients or donors, both of whom are comprised of racially diverse populations in the United States, is largely unknown.
Our observations in individuals with SCD receiving RhD, C, E, and K matched red cells from primarily Black donors suggests that RH variation on the part of both the blood donor and transfusion recipient contributes to unexpected Rh immunization. Approximately 85% of patients with SCD have at least 1 variant RH allele, although only 30% of unexpected Rh antibodies could be explained by homozygous inheritance of partial or altered RH alleles.9,12 Furthermore, 18% of Rh antibodies identified occurred in patients with the corresponding conventional RH allele.9 Examination of the RH genotypes of Black donors revealed a frequency of variant alleles that was similar to that found in patients with SCD, suggesting altered Rh epitopes on donor red cells may be contributing to unexplained Rh alloimmunization.12 Maintaining a CEK− inventory of units relies on Black donors, among whom CEK− phenotypes are more common compared with donors of Asian and European descent.8 Using the same CEK− donor inventory maintained primarily for patients with SCD may impact alloimmunization among patients with thalassemia receiving prophylactic CEK-matched transfusions.
The primary aim of this study was to examine alloimmunization among 40 chronically transfused patients with thalassemia of diverse races, including Asian/Indian, White, Black, or Middle Eastern who received serologic CEK-matched red cells. To better understand Rh immunization, we examined the RH genes among this patient cohort and assessed whether Rh antibodies were associated with inheritance of RH variants or with recent transfusion from Black donors who are known to have a high frequency of RH variants. Finally, we investigated how these factors contribute to Rh alloimmunization in comparison with a cohort of patients with SCD receiving chronic, simple transfusion therapy who were CEK-matched with Black donors.
Methods
Under an institutional review board–approved protocol, we retrospectively reviewed the clinical records (birth to 31 December 2018) of 40 patients with thalassemia and 48 patients with SCD who received chronic simple transfusion therapy at The Children’s Hospital of Philadelphia. Patients with thalassemia or SCD were diagnosed by β-globin sequencing or hemoglobin quantification. Inclusion criteria were receipt of a period of chronic transfusions at our institution within the 5 years up to the data cutoff date of 31 December 2018 and the ability to provide informed consent.
Per institutional blood bank protocol, all patients with a hemoglobinopathy were prophylactically matched for ABO, Rh (D, C, and E), and K; patients who lack the antigen received antigen-negative units that were hemoglobin S (HbS) negative, leukocyte reduced, and irradiated. Rh antigen matching did not include c and e. A 3-cell antibody screen was performed before each transfusion or when a new antibody or DHTR was suspected. In 2003, antibody identification transitioned from tube method to a gel-based method (Ortho Clinical Diagnostics, Raritan, NJ). Antibodies detected before transfusion at our institution were excluded from our analysis. Red cell antigen typing was performed by tube method, but starting in 2017, we obtained genotypes via the Human Erythrocyte Antigen BeadChip (Bioarray/Immucor, Warren, NJ). RH genotyping was performed with RHD and RHCE BeadChip arrays (Bioarray/Immucor), and polymerase chain reaction–based assays, as described previously.9,12 We obtained donor race and red cell phenotype data from the blood supplier for units transfused in the 3 visits before detection of their new antibody.
Results
Subjects
The study cohort included 40 patients with thalassemia (mean age, 15.2 years) requiring chronic transfusion: 32 β thalassemia (80%), 4 hemoglobin E/β thalassemia (10%), 3 hemoglobin H/constant spring (7.5%), and 1 hemoglobin Showa-Yakushiji/β thalassemia (2.5%) (Table 1). Sixteen self-identified as Asian (40%), 11 as White (27.5%), 5 as Black (12.5%), 4 as Indian (10%), and 4 as Middle Eastern (10%). Eight patients had a splenectomy (20%). No patient had reported a pregnancy. Collectively, 6977 red cell units were transfused to these 40 patients. The mean number of exposures per patient was 174 units, the median 131 units, and the range was 2 to 675 units. The mean length of transfusion therapy at our institution was 7.6 years, the median was 6.3 years, and the maximum was 26.1 years. All patients initiated chronic transfusion at our institution after June 1997 with the exception of 1 patient whose first transfusion was in November 1991. The mean age of first transfusion at our institution was 7.3 years. Sixteen patients had initiated transfusion therapy before establishing care at our institution. Among them, 3 individuals had formed antibodies before receiving care at our institution and included 1 anti-K, 1 anti-E, and 1 anti-V. Two patients had received transfusions at an outside institution while attending college, including 1 patient who is D+ and formed anti-D (T29).
Demographics of 40 chronically transfused patients with thalassemia
| Patient parameters | Number (%) |
|---|---|
| Mean age,y | 15.2 |
| Female | 22 (55) |
| Thalassemia subtype | |
| β thalassemia | 32 (80) |
| Hemoglobin E/β thalassemia | 4 (10) |
| Hemoglobin H/constant spring thalassemia | 3 (7.5) |
| Hemoglobin Showa-Yakushiji/β thalassemia | 1 (2.5) |
| Race | |
| Asian | 16 (40) |
| White | 11 (27.5) |
| Black | 5 (12.5) |
| Indian | 4 (10) |
| Middle Eastern | 4 (10) |
| Splenectomy | 8 (20) |
| Red cell exposures (units) | |
| Collective total | 6977 |
| Mean | 174.4 |
| Median | 130.5 |
| Range | 2-675 |
| Chronic transfusion therapy length,y | |
| Mean | 7.6 |
| Median | 6.3 |
| Range | 0.1-26.1 |
| Alloimmunization | |
| Patients alloimmunized | 13 (32.5) |
| Alloimmunization prevalence | (32.5) |
| Total alloantibodies detected* | 18 |
| Alloimmunization rate (per 100-unit exposures) | 0.26 |
| Patient parameters | Number (%) |
|---|---|
| Mean age,y | 15.2 |
| Female | 22 (55) |
| Thalassemia subtype | |
| β thalassemia | 32 (80) |
| Hemoglobin E/β thalassemia | 4 (10) |
| Hemoglobin H/constant spring thalassemia | 3 (7.5) |
| Hemoglobin Showa-Yakushiji/β thalassemia | 1 (2.5) |
| Race | |
| Asian | 16 (40) |
| White | 11 (27.5) |
| Black | 5 (12.5) |
| Indian | 4 (10) |
| Middle Eastern | 4 (10) |
| Splenectomy | 8 (20) |
| Red cell exposures (units) | |
| Collective total | 6977 |
| Mean | 174.4 |
| Median | 130.5 |
| Range | 2-675 |
| Chronic transfusion therapy length,y | |
| Mean | 7.6 |
| Median | 6.3 |
| Range | 0.1-26.1 |
| Alloimmunization | |
| Patients alloimmunized | 13 (32.5) |
| Alloimmunization prevalence | (32.5) |
| Total alloantibodies detected* | 18 |
| Alloimmunization rate (per 100-unit exposures) | 0.26 |
Alloantibodies detected before transfusion at our institution were excluded.
Antibodies identified in patients with thalassemia transfused with RhD, C, E, and K matched RBCs
Thirteen patients with thalassemia were alloimmunized while receiving prophylactic CEK-matched transfusions (32.5%), with 18 alloantibodies detected, and an alloimmunization rate of 0.26 alloantibodies per 100 units transfused. Eight of these patients (20%) had also formed warm autoantibodies, and an additional 3 patients (7.5%) had warm autoantibodies only. Despite prophylactic serologic CEK matching, antibodies directed against the Rh system were the most frequent (n = 13, 72.2%): 5 anti-D, 4 anti-C, 2 anti-E, 1 anti-e, and 1 anti-V (Figure 1A). Eight of these antibodies formed in patients who typed positive for the corresponding antigen, and 4 antibodies developed in individuals who were negative for the antigen and had received antigen-negative units. One anti-V was detected in a V− patient who received a V+ unit 3 months before. Additional non-Rh alloantibodies included the following: 1 anti-K, 1 anti-Kpa, 1 anti-Jsa, 1 anti-M, and 1 anti-S.
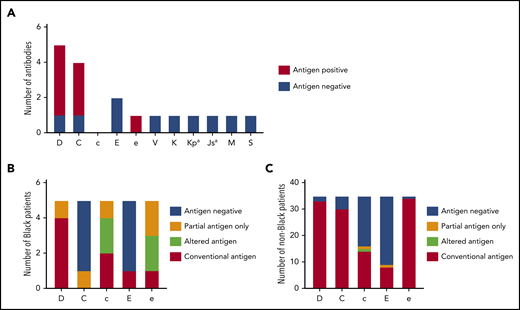
Alloimmunization and genotype-predicted Rh antigen expression among chronically transfused patients with thalassemia receiving prophylactic C, E, and Kmatched red cells. (A) Antibody specificities detected among 40 chronically transfused patients with thalassemia. Columns for each specificity indicate patients’ corresponding antigen status (positive or negative) as reported by standard serologic typing methods. RHD and RHCE genotype-predicted Rh antigen expression among 5 Black (B) and 35 non-Black (C) patients with thalassemia. Partial antigens predicted from genotypes associated with alleles that result in Rh epitope(s) missing and absence of conventional antigen. RHD*DAU0 or RHCE*ce48C has not been shown to encode Rh proteins lacking epitopes and is considered altered antigens.
Alloimmunization and genotype-predicted Rh antigen expression among chronically transfused patients with thalassemia receiving prophylactic C, E, and Kmatched red cells. (A) Antibody specificities detected among 40 chronically transfused patients with thalassemia. Columns for each specificity indicate patients’ corresponding antigen status (positive or negative) as reported by standard serologic typing methods. RHD and RHCE genotype-predicted Rh antigen expression among 5 Black (B) and 35 non-Black (C) patients with thalassemia. Partial antigens predicted from genotypes associated with alleles that result in Rh epitope(s) missing and absence of conventional antigen. RHD*DAU0 or RHCE*ce48C has not been shown to encode Rh proteins lacking epitopes and is considered altered antigens.
RH genetic diversity in patients with thalassemia
RH genotyping was performed to identify individuals who express partial Rh antigens and could be at risk for Rh alloantibody formation if exposed to the Rh epitopes they lack. Among the 40 patients, 10 had at least 1 variant RH allele: all 5 who self-identified as Black, 3 Asian, 1 Indian, and 1 White. Overall, 6 RHD (7.5%) and 11 RHCE (13.8%) alleles were altered (Table 2). As expected, most RH-altered alleles were detected among Black patients: 4 of 10 RHD (40%) and 8 of 10 RHCE (80%) alleles (Table 2). RH genotyping predicted that 4 of the Black individuals exclusively expressed a partial Rh antigen: 1 D+ patient with RHD*DAU3 and *DIVa, 1 C+ patient with partial C encoded by RHCE*CeCW, 1 c+e+ patient with partial c and e antigens encoded by homozygous RHCE*ce(733G), and 1 e+ patient with partial e encoded by RHCE*ce(733G) (Figure 1B). Altered c and e antigens were assigned to 2 Black patients with RHCE*ce(48C) and no conventional allele in trans. The RHCE*ce(48C) allele, which is common among Black individuals, has not been shown to lack Rh epitopes. This finding is in contrast to the 35 non-Black patients, among whom only 2 RHD [2.9%, RHD*D(48C), *DV type 1] and 3 RHCE (4.3%, 2 RHCE*ce48C and 1 *cE48C) alleles were variant (Table 2), and identified in 4 individuals who self-identified as Asian and 1 as White. Only 1 patient with RHCE*cE48C potentially expresses partial c and E, but the allele is very rare, and the clinical significance is unclear (Figure 1C).
RHD and RHCE alleles among 40 chronically transfused patients with thalassemia
| RHD allele | Black | Non-Black | RHCE allele | Black | Non-Black | ||||
|---|---|---|---|---|---|---|---|---|---|
| No. | Frequency | No. | Frequency | No. | Frequency | No. | Frequency | ||
| RHD conventional | 6 | 0.600 | 59 | 0.843 | Ce conventional | 0 | 0.000 | 48 | 0.686 |
| Deleted D | 0 | 0.000 | 9 | 0.129 | ce conventional | 1 | 0.100 | 10 | 0.143 |
| D(48C) | 0 | 0.000 | 1 | 0.014 | cE conventional | 1 | 0.100 | 8 | 0.114 |
| Weak D type 1 | 0 | 0.000 | 1 | 0.014 | CE conventional | 0 | 0.000 | 1 | 0.014 |
| DAU0 | 2 | 0.200 | 0 | 0.000 | ce48C | 3 | 0.300 | 2 | 0.029 |
| DAU3 | 1 | 0.100 | 0 | 0.000 | ce733G | 3 | 0.300 | 0 | 0.000 |
| DIVa | 1 | 0.100 | 0 | 0.000 | ceTI | 1 | 0.100 | 0 | 0.000 |
| cE48C | 0 | 0.000 | 1 | 0.014 | |||||
| CeCW | 1 | 0.100 | 0 | 0.000 | |||||
| RHD allele | Black | Non-Black | RHCE allele | Black | Non-Black | ||||
|---|---|---|---|---|---|---|---|---|---|
| No. | Frequency | No. | Frequency | No. | Frequency | No. | Frequency | ||
| RHD conventional | 6 | 0.600 | 59 | 0.843 | Ce conventional | 0 | 0.000 | 48 | 0.686 |
| Deleted D | 0 | 0.000 | 9 | 0.129 | ce conventional | 1 | 0.100 | 10 | 0.143 |
| D(48C) | 0 | 0.000 | 1 | 0.014 | cE conventional | 1 | 0.100 | 8 | 0.114 |
| Weak D type 1 | 0 | 0.000 | 1 | 0.014 | CE conventional | 0 | 0.000 | 1 | 0.014 |
| DAU0 | 2 | 0.200 | 0 | 0.000 | ce48C | 3 | 0.300 | 2 | 0.029 |
| DAU3 | 1 | 0.100 | 0 | 0.000 | ce733G | 3 | 0.300 | 0 | 0.000 |
| DIVa | 1 | 0.100 | 0 | 0.000 | ceTI | 1 | 0.100 | 0 | 0.000 |
| cE48C | 0 | 0.000 | 1 | 0.014 | |||||
| CeCW | 1 | 0.100 | 0 | 0.000 | |||||
Number and frequency of RH alleles among Black (n = 5) and non-Black (n = 35) individuals.
Patient and donor characteristics among Rh-alloimmunized patients with thalassemia
Table 3 shows the antibody specificity, RH genotypes and phenotype, patient race, and donor phenotype and race associated with unexplained Rh immunization in patients with thalassemia. Among the 13 Rh antibodies identified, the anti-V was identified in a V− individual who had been exposed to a V+ unit from a Black donor 10 weeks before antibody identification. The V antigen is considered a low-prevalence Rh antigen in most populations but occurs in 30% of Blacks.13 None of the remaining 12 Rh antibodies could be attributed to a variant RH genotype in the patient. Only 1 patient (T29) had a common altered allele that is not associated with partial antigen expression and is not homozygous (Table 3). Notably, 8 antibodies (4 anti-D, 3 anti-C, 1 anti-e) were detected in antigen-positive patients whose RH genotypes predicted expression of the conventional antigen, and 4 antibodies (1 anti-D, 1 anti-C, and 2 anti-E) were detected in antigen-negative patients who had received antigen-negative units (Table 3).
Patient and donor characteristics for 3 transfusions preceding detection of unexplained Rh antibody specificities in individuals with thalassemia
| UPID | Specificity | RH haplotypes (presumed) | Patient Rh phenotype | Patient race | Donor unit phenotypes | No. units from Black donors | |
|---|---|---|---|---|---|---|---|
| T7 | anti-D | RHD-Ce | DeletedD-ce | D+ C+ c+ E− e+ | Middle Eastern | 6D+, 2 D− | 5 of 8 |
| T12 | anti-C | RHD-Ce | RHD-CE | D+ C+ c− E+ e+ | White | 6C+, 0C− | 5 of 6 |
| anti-D | 6D+, 0D− | 5 of 6 | |||||
| T23 | anti-C | RHD-Ce | RHD-cE | D+ C+ c+ E+ e+ | Asian | 0C+, 6C− | 5 of 6 |
| T25 | anti-D | RHD-Ce | RHD-cE | D+ C+ c+ E+ e+ | Asian | 4D+, 2D− | 2 of 6 |
| T29* | anti-D | RHD-ce | RHD-ce(48C) | D+ C− c+ E− e+ | White | 5D+, 2D− | 3 of 7 |
| T30 | anti-E† | RHD-Ce | RHD-Ce | D+ C+ c− E− e+ | White | 0E+, 3E− | 3 of 3 |
| T34 | anti-E† | RHD-Ce | RHD-Ce | D+ C+ c− E− e+ | Indian | 0E+, 7E− | 4 of 7 |
| anti-C | 0C+, 6C− | 5 of 6 | |||||
| anti-e | 7e+, 0e− | 6 of 7 | |||||
| T52 | anti-C† | DeletedD-ce | DeletedD-ce | D− C− c+ E− e+ | Middle Eastern | 0C+, 3C− | 0 of 3 |
| anti-D† | 0D+, 5D− | 1 of 5 | |||||
| UPID | Specificity | RH haplotypes (presumed) | Patient Rh phenotype | Patient race | Donor unit phenotypes | No. units from Black donors | |
|---|---|---|---|---|---|---|---|
| T7 | anti-D | RHD-Ce | DeletedD-ce | D+ C+ c+ E− e+ | Middle Eastern | 6D+, 2 D− | 5 of 8 |
| T12 | anti-C | RHD-Ce | RHD-CE | D+ C+ c− E+ e+ | White | 6C+, 0C− | 5 of 6 |
| anti-D | 6D+, 0D− | 5 of 6 | |||||
| T23 | anti-C | RHD-Ce | RHD-cE | D+ C+ c+ E+ e+ | Asian | 0C+, 6C− | 5 of 6 |
| T25 | anti-D | RHD-Ce | RHD-cE | D+ C+ c+ E+ e+ | Asian | 4D+, 2D− | 2 of 6 |
| T29* | anti-D | RHD-ce | RHD-ce(48C) | D+ C− c+ E− e+ | White | 5D+, 2D− | 3 of 7 |
| T30 | anti-E† | RHD-Ce | RHD-Ce | D+ C+ c− E− e+ | White | 0E+, 3E− | 3 of 3 |
| T34 | anti-E† | RHD-Ce | RHD-Ce | D+ C+ c− E− e+ | Indian | 0E+, 7E− | 4 of 7 |
| anti-C | 0C+, 6C− | 5 of 6 | |||||
| anti-e | 7e+, 0e− | 6 of 7 | |||||
| T52 | anti-C† | DeletedD-ce | DeletedD-ce | D− C− c+ E− e+ | Middle Eastern | 0C+, 3C− | 0 of 3 |
| anti-D† | 0D+, 5D− | 1 of 5 | |||||
Patient Rh antigen phenotype was predicted by RHD and RHCE genotype.
T29 also received transfusions at an outside hospital that were CEK-matched while at college before anti-D.
†Denotes Rh specificities identified in antigen-negative patients who received antigen negative units. Antibodies listed in order of identification for patients with multiple specificities: UPID T12 anti-C 5/2012, -D 2/2015; T34 anti-E 5/2017, -11C/2017, -e 3/2018; T52 anti-C 4/2011, -D 2/2015, and also had anti-Jsa 7/2011, -Kpa 12/2015.
Across the 3 visits preceding each unexplained Rh alloimmunization event, 44 (62.9%) of the 70 red cell units transfused were from donors who self-identified as Black. For 11 of the 12 unexplained Rh alloantibodies, at least 1 unit in the 3 prior transfusions was from a Black donor (Table 3). The anti-C detected for patient T52 was the only alloimmunization event not preceded by an exposure from a Black donor in the 3 visits before antibody detection, likely because of the requirement for D-negative units that are much less frequent among Black donors.13 However, when we examined 6 visits before detection of the anti-C, 2 of the 4 additional units transfused were from Black donors (given within 5 months of antibody detection).
Patient and donor characteristics among Rh-alloimmunized patients with SCD receiving chronic simple transfusion
For comparison, we examined patient and donor characteristics for 18 Rh antibodies formed by patients with SCD (n = 48; mean age, 11.6 years; supplemental Table 1) receiving chronic simple transfusion with prophylactic CEK-matched red cells from our donor program that specifically recruits Black donors to meet their antigen-negative unit needs (Blue Tag Program). This cohort was selected because chronic transfusion therapy was administered by simple transfusion with 1 to 2 donor exposures per transfusion visit, similar to the patients with thalassemia. This is in contrast to 3 to 10 donor exposures per transfusion for patients with SCD undergoing chronic red cell exchange. Two anti-D, 6 anti-C, 2 anti-E, 6 anti-e, 1 anti-f, and 1 anti-hrs like specificities were identified (Figure 2A). The collective red cell exposure for this cohort was 2701 units, the median red cell exposure was 25 units, and the mean was 56.3 units (supplemental Table 1). Among this cohort, 40 RHD (41.7%) and 53 RHCE (55.2%) alleles were altered (supplemental Table 2). Eighteen of these patients (33.3%) exclusively expressed at least 1 partial Rh antigen, including 6 partial D, 2 partial C, 8 partial c, and 6 partial e antigens (Figure 2B). In addition, genotyping predicted 8 altered RHD*DAU0 without conventional RHD, 12 altered c, and 11 altered e because of RHCE*ce48C.
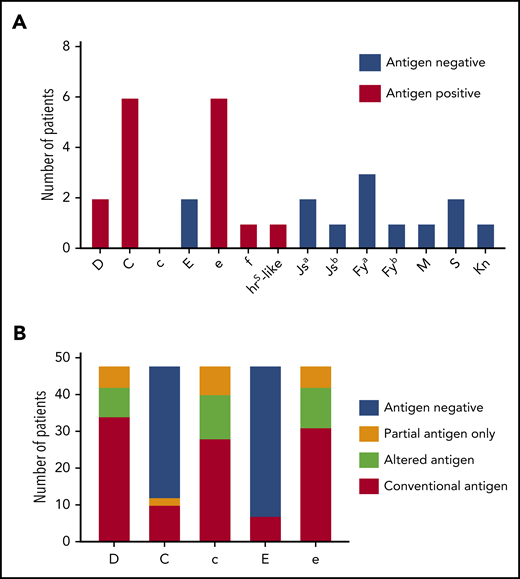
Alloimmunization and genotype-predicted Rh antigen expression among chronically transfused patients with SCD receiving prophylactic C, E, and Kmatched red cells by simple transfusion. (A) Antibody specificities detected among 48 chronically transfused patients with SCD. Columns for each specificity indicate patients’ corresponding antigen status (positive or negative) as reported by standard serologic or genotyping methods. (B) RHD and RHCE genotype-predicted Rh antigen expression among patients with SCD. Partial antigens predicted from genotypes with variant alleles that result in Rh epitope(s) missing and absence of conventional antigen. RHD*DAU0 or RHCE*ce48C has not been shown to encode Rh proteins lacking epitopes and is considered altered antigens.
Alloimmunization and genotype-predicted Rh antigen expression among chronically transfused patients with SCD receiving prophylactic C, E, and Kmatched red cells by simple transfusion. (A) Antibody specificities detected among 48 chronically transfused patients with SCD. Columns for each specificity indicate patients’ corresponding antigen status (positive or negative) as reported by standard serologic or genotyping methods. (B) RHD and RHCE genotype-predicted Rh antigen expression among patients with SCD. Partial antigens predicted from genotypes with variant alleles that result in Rh epitope(s) missing and absence of conventional antigen. RHD*DAU0 or RHCE*ce48C has not been shown to encode Rh proteins lacking epitopes and is considered altered antigens.
In contrast to the patients with thalassemia, inheritance of variant RH alleles encoding only the corresponding partial antigen could potentially explain 5 (27.8%) of the 18 Rh antibodies detected in this SCD cohort (Table 4; S205, S348 [anti-e and -f], S425, S775). One anti-E occurred in an E− patient (S348) who was exposed to E+ red cells after forming anti-e and then transfused with e-negative units in 2005. The hrS-like antibody was detected in Unique Patient Identifier S108, who is predicted to be hrS+ by RH genotype but had made a prior antibody identified as anti-e. The remaining 11 Rh antibodies were likely from exposure to Black donors with Rh variants. Seven antibodies (6 anti-C and 1 anti-E) were detected in antigen-negative patients who had received antigen-negative units (Table 4). Three antibodies (1 anti-D in S450 and 2 anti-e in S108 and S148) were identified in antigen-positive patients who are predicted to express the conventional antigen. Homozygosity for RHCE*ce(48C) in UPID S108 may provide a potential explanation for the anti-e, but this allele has not been associated with loss of Rh epitopes. Notably, several patients with SCD (UPID S108, S180, and S775) did not consistently receive units from Black donors once they became alloimmunized and had additional antigen-negative requirements. For example, 0 of 7 units for S108 at the time of anti-hrS–like detection and 0 of 6 red cell units for S775 at the time of anti-C detection were from Black donors, because e antigen-negative units were requested, which are relatively rare among Blacks.
Patient and donor characteristics for 3 transfusions preceding detection of Rh antibody specificities in individuals with SCD
| UPID | Specificity | RH haplotypes (presumed) | Patient predicted Rh phenotype | Donor unit phenotypes | Units from Black donors | |
|---|---|---|---|---|---|---|
| S 93 | anti-e | RHD-Ce | RHD-ce(733G) | D+ C+ cP E− e+ | 6e+, 0e− | 5 of 6 |
| S 108 | anti-e | DAU0-ce(48C) | Inactive RHD Ψ-ce(48C) | DA C− cA E− eA hrS+ | 4e+, 2e− | 4 of 6 |
| anti-hrS–like | Not typed for hrS | 0 of 7 (e− units) | ||||
| S 148 | anti-C†* | Weak partial 4.0-ce | Weak partial 4.0-ce(48C,733G) | DP C− c+ E− e+ | 0C+, 3C− | 3 of 3 |
| anti-e* | 3e+, 0e− | 3 of 3 | ||||
| S 159 | anti-E† | RHD-ce | DeletedD-ce(733G) | D+ C− c+ E− e+ | 0E+, 3E− | 3 of 3 |
| S 180 | anti-C† | RHD-ce(48C,733G) | DAU0-ce(48C) | D+ C− cA E− eA | 0C+, 4C− | 1 of 4 (Jsa− units) |
| S 205 | anti-D | Weak partial 4.0-ce(48C) | Inactive RHD Ψ-ce | DP C− c+ E− e+ | 4D+, 0D− | 3 of 4 |
| S 348 | anti-C† | RHD-ce(733G) | Weak partial 4.0-ce(48C,733G) | D+ C− cP E− eP fP | 0C+, 3C− | 1 of 3 |
| anti-e | 3e+, 0e− | 3 of 3 | ||||
| anti-E | 1E+, 2E− | 2 of 3 | ||||
| anti-f | Not typed for f | 2 of 6 | ||||
| S 425 | anti-e | RHD-cE | RHD-ce(733G) | D+ C- c+ E+ eP | 3e+, 0e− | 3 of 3 |
| S 450 | anti-D* | RHD-ce(733G) | DAU0-ce | D+ C- c+ E- e+ | 3D+, 0D− | 2 of 3 1 unknown |
| anti-C†* | 0C+, 3C− | 2 of 3 1 unknown | ||||
| S 775 | anti-e | RHD-cE | DIVa-ceTI | D+ C- c+ E- eP | 1e+, 2e− | 1 of 3 |
| anti-C† | 0C+, 6C− | 0 of 6 (e− units) | ||||
| S 1007 | anti-C† | RHD-ce(48C,733G) | RHD-ce | D+ C- c+ E- e+ | 0C+, 6C− | 4 of 6 |
| UPID | Specificity | RH haplotypes (presumed) | Patient predicted Rh phenotype | Donor unit phenotypes | Units from Black donors | |
|---|---|---|---|---|---|---|
| S 93 | anti-e | RHD-Ce | RHD-ce(733G) | D+ C+ cP E− e+ | 6e+, 0e− | 5 of 6 |
| S 108 | anti-e | DAU0-ce(48C) | Inactive RHD Ψ-ce(48C) | DA C− cA E− eA hrS+ | 4e+, 2e− | 4 of 6 |
| anti-hrS–like | Not typed for hrS | 0 of 7 (e− units) | ||||
| S 148 | anti-C†* | Weak partial 4.0-ce | Weak partial 4.0-ce(48C,733G) | DP C− c+ E− e+ | 0C+, 3C− | 3 of 3 |
| anti-e* | 3e+, 0e− | 3 of 3 | ||||
| S 159 | anti-E† | RHD-ce | DeletedD-ce(733G) | D+ C− c+ E− e+ | 0E+, 3E− | 3 of 3 |
| S 180 | anti-C† | RHD-ce(48C,733G) | DAU0-ce(48C) | D+ C− cA E− eA | 0C+, 4C− | 1 of 4 (Jsa− units) |
| S 205 | anti-D | Weak partial 4.0-ce(48C) | Inactive RHD Ψ-ce | DP C− c+ E− e+ | 4D+, 0D− | 3 of 4 |
| S 348 | anti-C† | RHD-ce(733G) | Weak partial 4.0-ce(48C,733G) | D+ C− cP E− eP fP | 0C+, 3C− | 1 of 3 |
| anti-e | 3e+, 0e− | 3 of 3 | ||||
| anti-E | 1E+, 2E− | 2 of 3 | ||||
| anti-f | Not typed for f | 2 of 6 | ||||
| S 425 | anti-e | RHD-cE | RHD-ce(733G) | D+ C- c+ E+ eP | 3e+, 0e− | 3 of 3 |
| S 450 | anti-D* | RHD-ce(733G) | DAU0-ce | D+ C- c+ E- e+ | 3D+, 0D− | 2 of 3 1 unknown |
| anti-C†* | 0C+, 3C− | 2 of 3 1 unknown | ||||
| S 775 | anti-e | RHD-cE | DIVa-ceTI | D+ C- c+ E- eP | 1e+, 2e− | 1 of 3 |
| anti-C† | 0C+, 6C− | 0 of 6 (e− units) | ||||
| S 1007 | anti-C† | RHD-ce(48C,733G) | RHD-ce | D+ C- c+ E- e+ | 0C+, 6C− | 4 of 6 |
Patient Rh antigen phenotype was predicted by RHD and RHCE genotype. Partial (P) antigens predicted from genotypes with variant alleles that result in Rh epitope(s) missing and absence of conventional antigen. Altered (A) predicted for patients with RHD*DAU0 or RHCE*ce48C and no conventional allele in trans as these 2 alleles have not been shown to lack epitopes. Antibody specificities in bold are potentially explained by patient’s RH genotype. Antibodies listed in order of identification for patients with multiple specificities: UPID S108 anti-e 12/2009, -hrS-like 3/2010; S148 anti-C and -e 5/2007; S348 anti-C 8/2004, -e 7/2005, -E 9/2005, -f 4/2014; S450 anti-D and -C 5/2005; S205 anti-C 8/2004, -D 7/2018, S775 anti-e 5/2013, -C 9/2014.
Two antibodies detected on the same date.
Denotes Rh specificities identified in antigen-negative patients who received antigen-negative units.
Discussion
We report the first analysis of alloimmunization among 40 individuals with thalassemia of diverse racial backgrounds receiving chronic transfusions with prophylactic Rh (D, C, E) and K matched red cells. Reported alloimmunization prevalence ranges from 3% to 42%, and it has been suggested that lower prevalence correlates with population homogeneity.2,5 We observed an alloimmunization prevalence of 32.5% among a cohort of chronically transfused patients with a median red cell transfusion burden of 131 units (37.5% prevalence if antibodies formed before care at our institution are included). The rate of 0.26 alloantibodies/100 units transfused is similar to that for patients with SCD receiving prophylactic CEK-matched red cells at our institution and others (0.26-0.5 alloantibodies per 100 units transfused).9,14,15
This study confirms the high proportion of individuals that identify as Black having at least 1 variant RH allele: 5 (100%) of the 5 Black patients with thalassemia and 38 (88.4%) of the 43 Black patients with SCD. This finding is in contrast to the 35 non-Black patients with thalassemia, among whom only 5 (14.3%) had a variant RH allele. Among these 5 patients, 4 are Asian, and the remaining individual self-identified as White but carries an allele common in Blacks (RHCE*ce48C), reflecting the racial admixture in the United States. Because this retrospective study spans more than 15 years of patient transfusion history, we were unable to RH genotype the donors implicated in alloimmunization events to confirm variant Rh exposures.
In contrast to 28% of Rh antibodies in individuals with SCD that can be explained by partial Rh antigen expression on their red cells, inheritance of variant RH alleles did not explain any of the Rh antibodies formed in patients with thalassemia. In fact, among all 8 patients who are antigen positive and formed the corresponding antibody, RH genotyping revealed corresponding conventional alleles only. These observations suggest that donor RH variation may be an underappreciated risk factor for alloimmunization. We previously reported that, among 1444 patients with SCD and healthy Black donors, 29% of RHD and 53% of RHCE alleles were altered.12 Thus, Rh variants are common among Black donors, and nearly all patients with thalassemia or SCD had received red cells from Black donors in the 3 transfusions preceding new, unexplained Rh antibody specificities. A patient exposed to donor red cells with variant Rh antigens may recognize these as foreign and form an alloantibody. If the patient types antigen positive, these may be classified as autoantibodies but are likely alloantibody responses to complex foreign Rh epitopes on the donor red cells. Some unexplained Rh antibodies may also be caused by Rh mimicking or cross-reactive epitopes, which include expression of C-like epitopes on RhD, D-like epitopes on RhCe, and E-like epitopes on RhCe proteins,16-18 adding additional complexity to the serologic findings.
Because the prevalence of red cell antigens is heavily influenced by racial origin, recruiting Black donors for patients with SCD may offer additional antigen compatibility for FY, JK, MNS, and other blood group antigens. For non-Black patients with thalassemia, the use of Black donors does not confer this same advantage and may inadvertently expose patients to immunogenic Rh epitopes not detected by standard typing. Units lacking both C and E, as well as K, are significantly more prevalent in donors of African ancestry compared with those of European descent, which may lead blood centers to selectively antigen type minority donors to support CEK-matching programs in the United States. An unintended consequence of this practice may be the higher rate of Rh alloimmunization because of donor RH variation if matching is limited to C and E only. In this study, 15 Asian/Indian and 5 White patients with thalassemia were C+c−, and none had formed anti-c, including 6 alloimmunized patients whose donor phenotypes retrospectively confirm exposure to c+ units. However, because many thalassemia patients are of Asian ancestry and have a high prevalence of DCe (R1) haplotypes, extending matching for c, and rarely e, could avoid use of Black donor units (typically c+e+), which should be reserved for patients with SCD. As 19 of the 35 non-Black patients with thalassemia lack c and 1 lacks e, implementation of Rh C, c, E, and e matching would divert these requests to the general donor inventory and improve CEK unit inventory management. Further non-Rh antigen mismatch may also result in alloimmunization, as we observed in the 1 anti-Kpa and 1 anti-Jsa formed by a non-Black patient with thalassemia. Both of these antigens have a higher prevalence among individuals of African descent.
Only 1 male patient, T52 who is C−, had formed an unexplained anti-C and had no Black donor exposure identified in the preceding three transfusion visits. He had been transfused with 2 Black donor units within 5 months of anti-C detection; reaching the level of antibody detection can take more than 3 months.19 This individual is D− and also formed anti-D while receiving D− units, for which only 1 of the 5 units transfused in the prior 3 visits was from a Black donor. Anti-G in a D−C− individual was not considered, given that the anti-D was identified nearly 4 years after the anti-C. It is also unlikely that the patient was exposed to a weak D unit that was mislabeled D negative, because the anti-D was not persistently detectable. It was detected in 2 specimens only: at initial identification and 10 months later. We excluded transfusion outside our institution and donor red cell mistyping by retyping segments from donor units in the 2 preceding transfusions when the unexpected Rh antibodies were identified. We also identified 1 anti-K in an Asian patient that was unexplained. The patient typed K−, retained segments from the units transfused in the prior 2 visits were retyped as K−, and the patient had not been transfused outside our institution.
Similar to Rh antibodies observed in patients with SCD,9 most antibodies had transient demonstration in the patient’s plasma. Antibody detection tests are performed regularly in these patients requiring chronic transfusion. Nine of the 12 unexplained Rh antibodies were detected only once or up to 6 months. UPID T52’s anti-D and -C were detectable for 10 and 59 months, respectively, and UPID T30’s anti-E was detectable for 43 months after first detection, but all became undetectable. Once identified, our institutional practice is to provide antigen-negative units for all future transfusions, even if the patient types antigen positive, and the antibody is no longer demonstrable. The 1 exception to this practice is anti-e in an individual who also lacks the E antigen, where decisions are made on a case-by-case basis.
Among the 13 Rh antibodies formed, none were associated with a clinical presentation of a DHTR. Review of the patient’s hemoglobin at the time of antibody detection in comparison with their pretransfusion hemoglobin levels in the preceding 6 months revealed 1 individual with a hemoglobin less than 1 g/dL below their pretransfusion baseline. At the time of anti-D detection, this D+ individual (T29) with conventional RHD alleles had a pretransfusion hemoglobin of 8.2 g/dL compared with her mean pretransfusion value of 9.9 g/dL from the preceding 8 visits. The 1 anti-Kpa was associated with a DHTR; the hemoglobin at time of antibody detection was 5.9 g/dL in an individual whose average pretransfusion hemoglobin was 8.8 g/dL. Although none of the Rh antibodies caused a DHTR, it is not possible to measure a subtle decline in transfused red cell survival or to determine the impact of additional antigen-negative requirements and transfusion delays while antibody identification is ongoing. For patients who are chronically transfused, these are not trivial considerations.
Among chronically transfused patients with thalassemia, the Rh antibodies identified in individuals whose own RH alleles do not explain the immunization underscore the need for more precise typing and matching of donor red cells. To mitigate Rh alloimmunization that occurs despite transfusions that appear matched based on serologic typing, extending Rh D, C, and E matching to include c and e would result in better-matched units and minimize exposure to Black donors who are likely to express Rh variants. This would also reserve the Black donor units for patients with SCD who rely on this valuable donor population. In the future, RH genotype-matched red cells may be valuable. Red cell genotyping has become more widely available, but the cost and expertise required for comprehensive RH genotyping to identify Rh variants currently prohibits widespread use. With advances in sequencing technology, we are optimistic that cost-effective genetic red cell matching will become available for chronically transfused patients with hemoglobinopathies, who appear particularly vulnerable to alloimmunization.
For data, please email the corresponding author at chous@email.chop.edu.
Acknowledgments
The authors thank the patients and families who enrolled in the studies, blood donors, and members of the CHOP Blood Bank, the Immunohematology and Genomics Laboratory at the New York Blood Center, and the donor center at the Penn-Jersey American Red Cross.
This work was supported by the Cooley’s Anemia Foundation Clinical Research in Thalassemia Grant (S.T.C.), National Institutes of Health, National Heart, Lung, and Blood Institute grant HL147879-01 (S.T.C. and C.M.W.), and a generous donation from the DiGaetano family.
Authorship
Contribution: S.J.W. and S.T.C. designed the study, analyzed results, and wrote the manuscript; S.U. obtained informed consent and maintained all clinical data in a research database; D.K., C.F., and S.V. conducted research and edited the manuscript; and D.F.F. and C.M.W. conducted research, analyzed results, and edited the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Stella T. Chou, Department of Pediatrics, The Children's Hospital of Philadelphia, 3615 Civic Center Blvd, Abramson Research Center, Room 316D, Philadelphia, PA 19104. e-mail: chous@email.chop.edu

