

Key Points
Fibrin-targeting nanogels promote maturation of actively polymerizing clots; when loaded with tPA, they lyse clots.
Loaded nanogels can treat microthrombi formation and bleeding complications associated with DIC.

Abstract
Disseminated intravascular coagulation (DIC) is a pathological coagulopathy associated with infection that increases mortality. In DIC, excessive thrombin generation causes symptoms from formation of microthrombi to multiorgan failure; bleeding risks can also be a concern because of clotting factor consumption. Different clinical events lead to DIC, including sepsis, trauma, and shock. Treatments for thrombotic episodes or bleeding presentation in DIC oppose each other, thus creating therapeutic dilemmas in management. The objective of this study was to develop fibrin-specific core-shell nanogels (FSNs) loaded with tissue-type plasminogen activator (tPA) to treat the microcirculatory complications of DIC, which would facilitate targeted clot dissolution to manage microthrombi and the potential consumptive coagulopathy that causes bleeding. FSNs enhance formation of actively polymerizing clots by crosslinking fibrin fibers, but they can also target preexisting microthrombi and, when loaded with tPA, facilitate targeted delivery to lyse the microthrombi. We hypothesized that this dual action would simultaneously address bleeding and microthrombi with DIC to improve outcomes. In vivo, tPA-FSNs decreased the presentation of multiorgan microthrombi, recovered platelet counts, and improved bleeding outcomes in a DIC rodent model. When incorporated with human DIC patient plasma, tPA-FSNs restored clot structure and clot growth under flow. Together, these data demonstrate that a fibrinolytic agent loaded into fibrin-targeting nanogels could improve DIC outcomes.
Introduction
Disseminated intravascular coagulation (DIC) is a condition characterized by pathological coagulopathy. It is associated with up to 19% of admissions to the intensive care unit and has a high mortality rate of 40% to 78%, depending on diagnostic criteria.1-4 Various clinical events can lead to DIC, including infection, trauma, cancer, pregnancy, and liver disease.5-11 DIC may occur in 30% to 50% of cases of severe sepsis, a leading cause of death worldwide,12-14 and also in critically ill patients with COVID-19 because of secondary infections.8-11 Novel therapies are critically needed to improve outcomes because of the high mortality associated with DIC.
DIC is a complex, acquired coagulopathy that is characterized by uncontrolled thrombin generation throughout the intravascular system.15,16 As thrombin is generated, fibrin deposits in the microvasculature, which leads to ischemic tissue damage and multiorgan failure. As part of this complex response, microthrombi consume clotting factors and can subsequently initiate fibrinolytic pathways that, in turn, lead to diffuse bleeding.15,17 Current treatment of DIC mainly involves treatment of the underlying condition, but in circumstances of severe thrombosis or severe bleeding, thrombolytic therapies or procoagulants, platelets, or plasma transfusions may be administered.7,14,18,19 However, because thrombosis and bleeding can occur simultaneously in DIC, treatment strategies for either presentation oppose each other. Controlling the hemostatic imbalance can be elusive, thus resulting in the significant mortality rate of DIC.
The coagulation cascade culminates in the formation of fibrin through the polymerization of fibrinogen that is initiated by the serine protease thrombin, forming an insoluble matrix.20 The dynamics of fibrin polymerization therefore play a central role in thrombosis and bleeding because thrombosis resulting from fibrin formation in the microcirculation, as in DIC, decreases blood flow, which causes tissue damage, whereas deficient fibrin polymerization is critical during hemostasis to prevent exsanguination.21-25 Methods of regulating the dynamics of fibrin polymerization include inhibiting the activation of coagulation by antithrombin, protein C, or tissue factor pathway inhibitor, most of which have been studied for treating thromboinflammatory diseases such as DIC.26,27 In addition, the fibrinolytic system regulates fibrin dynamics by lysing preformed clots that contain fibrin through plasmin activation.28 Tissue-type plasminogen activator (tPA) is a major initiator of fibrinolytic pathways because it catalyzes the conversion of plasminogen into plasmin to lyse clots.29 In DIC, deranged fibrin polymerization occurs for thrombi formation, along with a consumptive coagulopathy with inadequate substrates necessary for clot formation thereby leading to bleeding complications.
Targeting fibrin is a promising strategy in the development of therapeutics for thrombotic and/or hemostatic complications. However, with current treatment strategies for thrombosis and bleeding, off-target effects with systemic agents can easily alter the hemostatic balance at noninjured sites.30 Therefore, in treating a disorder such as DIC in which the coagulation imbalance occurs systemically, targeting a coagulation protein such as fibrin could help regulate excessive thrombi formation and bleeding risks. We hypothesize that simultaneously addressing bleeding and microthrombi associated with DIC will improve clinical outcomes. In this study, novel fibrin-specific nanogels (FSNs) have been developed that use nanoscale colloidal hydrogels coupled to a fibrin-specific antibody and are loaded with a fibrinolytic agent, tPA, to create a novel therapeutic that balances the procoagulant and fibrinolytic shutdown that occurs in DIC (Figure 1). Unloaded FSNs target the clotting process by crosslinking fibrin fibers and assist in maturation of actively polymerizing clots, whereas tPA-loaded nanogels facilitate directed lysis of aberrant thrombi formed in DIC (Figure 1A). The overall objective in this study was to examine the ability of fibrin-targeting core-shell nanogels to modulate overall fibrin dynamics in DIC by promoting lysis of preexisting clots and promoting clotting abilities at sites of injury to restore the overall hemostatic balance. We demonstrate in vivo in a rodent model of DIC and in vitro in DIC patient plasma samples that fibrin-targeting nanogels delivering tPA can treat aberrant clot formation to restore coagulation potential and mitigate DIC complications of thrombosis and bleeding to improve outcomes.
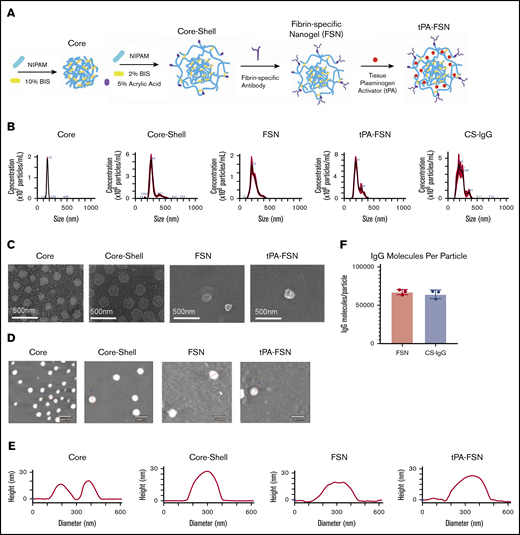
Nanogel synthesis schematic and characterization. (A) Schematic of core-shell particle synthesis and FSN fabrication followed by subsequent tPA loading through a rehydration technique. (B) Core, core-shell, FSN, tPA-FSN, and tPA-CS-IgG size characterization hydrodynamic measurements using 108 particles. (C) SEM (JEOL JSM-7600F) images are shown at a magnification of 80 000× to validate core-shell architecture and FSN and tPA-FSN structure. Representative AFM (Asylum Research MFP-3D) dry images using 30 particles (D) and representative height traces using 30 particles (E). (F) Quantification of antibody molecules per particle for FSN and CS-IgG particles. Data are presented as average ± standard deviation.
Nanogel synthesis schematic and characterization. (A) Schematic of core-shell particle synthesis and FSN fabrication followed by subsequent tPA loading through a rehydration technique. (B) Core, core-shell, FSN, tPA-FSN, and tPA-CS-IgG size characterization hydrodynamic measurements using 108 particles. (C) SEM (JEOL JSM-7600F) images are shown at a magnification of 80 000× to validate core-shell architecture and FSN and tPA-FSN structure. Representative AFM (Asylum Research MFP-3D) dry images using 30 particles (D) and representative height traces using 30 particles (E). (F) Quantification of antibody molecules per particle for FSN and CS-IgG particles. Data are presented as average ± standard deviation.
Methods
Nanogel synthesis and characterization
Core-shell poly(N-isopropylacrylamide) p(NIPAM) nanogels were synthesized in 2 precipitation polymerization reactions.31 Cores contained 90% NIPAM and 10% N,N′-methylenebis(acrylamide) (BIS) (Sigma-Aldrich), and shells containing 93% NIPAM, 2% BIS, and 5% acrylic acid (AAc) were synthesized around cores. AAc dispersed throughout the nanogel shell acts as chemoligation sites for subsequent antibody conjugation. Nanogels were filtered through glass wool and purified via dialysis. Methacryloxyethyl thiocarbamoyl rhodamine B monomer (0.1%) (PolyScience) was incorporated for fluorescence. For biodistribution studies, FSNs were conjugated to VivoTag-S 750 Fluorochrome (PerkinElmer). Single-layer nanogels were synthesized and contained 93% NIPAM, 2% BIS, and 5% AAc. Size and morphologic characterization included particle tracking analysis (Malvern Panalytical NanoSight), atomic force microscopy (AFM), and scanning electron microscopy (SEM). FSNs and control particles were made by conjugating either a sheep anti-human fibrin fragment E polyclonal antibody (Affinity Biologicals) or a sheep immunoglobulin G (IgG) isotype control (Thermo Fisher Scientific), respectively, to core-shell particles using 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide/sulfo N-hydroxysuccinimide (EDC/sulfoNHS) chemistry. From a known particle per mL concentration found through NanoSight particle tracking, the amount of fragment E antibody or control sheep IgG was determined by using a CBQCA Protein Quantitation Kit (Thermo Fisher Scientific) conducted in duplicate for 3 particle suspensions.
Nanogel drug loading and release characterization
Lyophilized particles were resuspended at high concentrations (20 mg/mL) in loading solutions of 29 μg/mL tPA (Sigma-Aldrich) to load through a rehydration technique, which allows for drug entanglement within the polymer network of the nanogels upon nanogel swelling in solution.32,33 After agitation for 24 hours, particles were centrifuged, washed, and resuspended in N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid (HEPES) buffer (25 mM HEPES, 0.15 M NaCl, 5 mM CaCl2 [pH 7.4]). At various time points (1-168 hours), the particles were centrifuged, and release supernatant was collected. Detection of released material was determined through fluorescence scans (for drug mimics) or tPA enzyme-linked immunosorbent assay (ELISA) (ab190812). Three separate samples for each condition were examined.
To examine potential aggregation of particles, FSNs and tPA-FSNs were added to platelet-poor plasma (PPP) (New York Blood Center), whole blood obtained in sodium citrate from lipopolysaccharide (LPS)–induced DIC rats, and whole blood with the addition of 10% rat complement serum (ab155157; Abcam). A final 2 mg/mL concentration of particles was added to plasma or whole blood samples with 25 μg/mL anti-sheep fluorescein isothiocyanate secondary antibody (Thermo Fisher Scientific), 5 mM CaCl2, 0.1 mg/mL AlexaFluor 594 conjugate fibrinogen (Thermo Fisher Scientific), and 0.5 U/mL thrombin. After 2 hours, confocal microscopy was used to visualize the particles.
Clotting analysis with tPA-loaded FSNs under static and fluidic conditions under active coagulation
tPA (29 μg/mL) was loaded into purified FSNs (tPA-FSNs) or CS-IgGs (tPA-CS-IgG). After separating loading solution, washing, and lyophilizing, particles were incorporated into an endogenous fibrinolysis absorbance–based clotting assay for static clotting analysis.34 To test clotting dynamics under fluidic conditions, a custom T-junction polydimethylsiloxane fluidic device was used. A stationary fibrin clot boundary was formed at the T junction, and actively polymerizing solutions were flowed into the device. Treatment conditions in the flow solution included buffer, FSNs, nonbinding core-shell nanogels (C/S), tPA-FSNs, tPA loaded into nonbinding core-shell nanogels (tPA-C/S), tPA alone, and tPA-CS-IgGs. Doses for tPA alone were previously established from studies using the same nanogel formulation.31 A negative control of FSNs under nonpolymerizing conditions was also conducted. Images were taken and the distance between clot apexes was measured for 20 minutes.
To examine how unloaded FSNs enhance fibrin polymerization, confocal microscopy was used to examine fibrin clot structure. Clots were made by adding 0.25 U/mL human α-thrombin (Thermo Fisher Scientific) and 0.05 mg/mL Alexa-Fluor 488–labeled fibrinogen (Thermo Fisher Scientific) to 2 mg/mL fibrinogen (FIB3, Enzyme Research Laboratories) in HEPES buffer with 1 mg/mL FSNs or CS-IgG control particles. Fiber density was quantified from 3 z-stacks per clot, and 3 separate clots were analyzed.
In vivo evaluation of tPA-FSN treatment and biodistribution in a rodent model of DIC
In vivo experimentation was approved by North Carolina State University Institutional Animal Care and Use Committee and conducted in Association for Assessment and Accreditation of Laboratory Animal Care International–accredited facilities. Adult male Sprague Dawley rats received intravenous infusions of 30 mg/kg LPS over 4 hours to induce DIC.35,36 Saline, FSNs, tPA-FSNs, or tPA-CS-IgGs (10 mg/kg) were then injected via tail vein and circulated for 30 minutes. Terminal blood draws were performed to evaluate coagulation parameters, and organs were harvested to evaluate thrombus formations. Control animals did not receive LPS or treatment. Martius scarlet blue (MSB) staining and immunohistochemistry (IHC) with anti-fibrin antibody (UC45) (GeneTex) were performed for visualizing fibrin microthrombi. Quantification of fibrin microthrombi was conducted from fibrin IHC tissue sections. Three tissue sections from the heart, lung, kidney, and liver from each animal were imaged using the EVOS FL Auto Imaging System. A particle count for microthrombi was obtained using ImageJ particle analysis software. Average fibrin particle counts for each organ for each animal are shown in Figure 3B-E.
Manual platelet counts were conducted using a hemocytometer, and PPP was isolated from whole blood collected in sodium citrate from each animal. Fibrinogen and D-dimer ELISAs were performed (Abcam, MyBioSource). Confocal and cryogenic SEM (cryoSEM) were used to examine ex vivo fibrin clot structure of PPP from control and DIC animals to further assess the clotting factor consumption of DIC. For confocal microscopy, plasma was polymerized by adding 0.5 U/mL human α-thrombin (Thermo Fisher Scientific) with 5 mM CaCl2 and 0.05 mg/mL Alexa-Fluor 488–labeled fibrinogen (Thermo Fisher Scientific) for visualization. Ex vivo clots polymerized for 2 hours before imaging. Fiber density was quantified from 3 z-stacks per clot. For cryoSEM, clots were made from PPP and 0.5 U/mL thrombin and were imaged 3 hours after polymerization.
To determine potential effects of circulating particles left in the plasma, a dot blot was performed on the plasma samples to detect particles. Rabbit anti-sheep IgG (31627; Thermo Fisher Scientific) was used as a primary antibody and IRDye 800CW goat anti-rabbit IgG secondary antibody was used for detection on a Licor Odyssey CLx Imaging system where subsequent analysis was conducted. In addition, 1 mg/mL of FSNs or tPA-FSNs was exogenously added to PPP clots made from DIC animals treated with saline. Clots were polymerized and imaged as described above with 1 U/mL thrombin. Fiber density was quantified as previously described to evaluate clot structure.
Biodistribution of tPA-FSNs and FSNs in DIC animals was conducted by injecting 10 mg/kg particles or saline after DIC induction with LPS, as described above. After 30 minutes, animals were euthanized, and organs, blood, and urine were collected. An IVIS Xenogen In Vivo Imager was used to scan organs ex vivo and evaluate the presence of particles in the heart, lungs, liver, kidneys, and spleen. A dot blot was performed on the urine and whole blood samples as described above.
In vivo evaluation of bleeding in a rodent model of DIC with tPA-FSN treatment
The LPS-DIC model was used, and after 30 minutes of treatment (saline, FSNs, tPA-FSNs, or tPA-CS-IgGs), the liver was injured (lacerated).37 Separate cohorts of animals acted as a control or vehicle condition. Blood loss was monitored for 10 minutes via blood collected in preweighed gauze held adjacent to the injury; before the cut was made, a ruler was used to measure a 3 × 1.5-cm line through the left lobe of the liver. Gauze was changed every 10 seconds for 30 seconds, every 30 seconds until 3 minutes, and every minute until 10 minutes and weighed immediately to quantify blood loss over time. Terminal cardiac puncture and organ/wound harvest was conducted. Platelet count, PPP isolation, and a fibrinogen ELISA were performed. MSB and IHC were performed on wound sections for fibrin (UC45) (GeneTex) and platelets (SJ19-09; Novus Biologicals). Confocal microscopy was used to examine fibrin clot structure of PPP and quantify fiber density from animals after injury, as above.
DIC patient plasma clot structure
DIC patient plasma was obtained from Duke University Medical Center with approval from the institutional review board and informed consent from patients. Clots formed from the plasma of 3 DIC patients were examined using confocal microscopy to visualize fibrin network properties, quantify fiber density, and compare with normal human plasma clots. Clots from PPP were made with 1 U/mL thrombin and 0.05 mg/mL Alexa-Fluor 488–labeled fibrinogen in HEPES buffer. FSNs, tPA-FSNs, or tPA-CS-IgGs (1 mg/mL) were incorporated into DIC patient plasma clots before polymerization. Healthy adult human PPP used for control was obtained from the New York Blood Center (New York, NY). Two duplicate clots were analyzed for each sample per condition, imaging and analyzing 3 images per duplicate.
Evaluation of clotting dynamics of DIC patient plasma in a microfluidic device under active coagulation
A custom-made Y-shaped microfluidic device was fabricated by soft lithography with a channel cross-section of 500 × 234 µm. A stationary fibrin clot boundary was formed at the Y junction with DIC patient plasma. Actively polymerizing flow solutions were flowed into the device containing corresponding DIC patient plasma for 20 minutes at wall shear rates of 10 s−1. Images were taken over 20 minutes, and clot boundary growth at the Y junction was quantified.
Statistical analysis
Statistical analysis was performed by using GraphPad Prism 8 (GraphPad, San Diego, CA). Dextran release data were analyzed using multiple 2-tailed Student t tests correcting for multiple comparisons using the Holm-Sidak method with α = 0.05. Other data sets were analyzed via a 1-way analysis of variance with a Tukey’s post hoc test using a 95% confidence interval. Outlier tests were performed on all data sets before statistical analysis. All data are presented as average ± standard deviation.
Results
Core-shell nanogels offer burst release profiles
Nanogels offer a tunable platform for drug delivery based on polymer network and crosslinking.33 p(NIPAM) specifically allows for complex architectures for customizable drug delivery.38 We have previously shown that core-shell nanogel architectures with highly crosslinked cores facilitate partitioning of protein therapeutics into looser crosslinked shells for rapid release; we therefore used core-shell architecture here, comparing release dynamics to a single-layer nanogel.31 Hydrodynamic core size, AFM diameter, and height measurements increased from 170 ± 17 nm, 166 ± 16 nm, and 17 ± 3 nm, respectively, to 287 ± 67 nm, 263 ± 28 nm, and 29 ± 4 nm upon shell addition. SEM images further validate shell addition (Figure 1). Hydrodynamic diameter, AFM diameter, and height measurements for FSNs are similar to those for unconjugated counterparts (239 ± 55 nm, 254 ± 61 nm, and 25 ± 10 nm) and for tPA-FSNs (225 ± 53 nm, 273 ± 43 nm, and 29 ± 12 nm). Hydrodynamic diameter for CS-IgG particles shows size similar to that of fibrin-specific counterparts at 225 ± 61 nm. FSNs display 67 200 ± 3380 fibrin fragment E antibody molecules per particle. CS-IgG particles display 64 200 ± 5580 sheep IgG isotype control molecules per particle.
To compare drug release, particle analysis verified that single-layer 2% BIS crosslinked particles were similar in size to core-shell particles (268 ± 34 nm). When loaded with 70 kDa fluorescent dextran, mimicking tPA, releasate samples showed that core-shell particles offer a more burst release profile compared with single-layer nanogels such that a significantly larger percent of payload is delivered at early time points (supplemental Figure 1). With applications for DIC for which ischemic damage is a concern, burst release profiles are desired. Therefore, subsequent studies used core-shell particles. Once conjugated to a sheep anti-human fibrin fragment E antibody or a sheep IgG isotype control to make FSNs and CS-IgG particles, respectively, tPA loading efficacy into the particles was determined to be 51% ± 8.5% for FSN and 46% ± 7% for CS-IgG (Figure 2A). Both FSN and CS-IgG particle types displayed burst release profiles over 44 hours (Figure 2B). In addition, both FSNs and tPA-FSNs added to plasma, whole blood, and whole blood with 10% added complement serum did not show substantial particle aggregation (supplemental Figure 2).
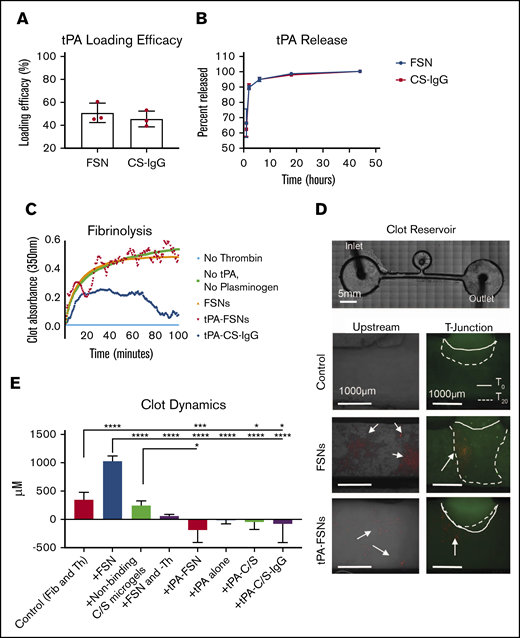
In vitro evaluation of drug release characteristics and clotting under static and dynamic conditions. (A) tPA loading efficacy comparing FSN and CS-IgG particles (n = 3 nanogel samples). (B) tPA release studies comparing FSN and CS-IgG particles (n = 3 nanogel samples). (C) Absorbance-based polymerization-degradation assay with tPA loaded particles (n = 3 clots per condition). (D) In vitro clotting under dynamic conditions in a fluidic device that contained a stationary fibrin clot located in the clot reservoir and that used flow solutions with fibrinogen and thrombin (control) (n = 7 replicates), fibrinogen and thrombin with unloaded FSNs (n = 4 replicates), fibrinogen and thrombin with nonbinding core-shell (CS) nanogels (n = 3 replicates), fibrinogen with unloaded FSNs without thrombin and thus no active coagulation (n = 3 replicates), fibrinogen and thrombin with tPA-FSNs (n = 4 replicates), fibrinogen and thrombin with tPA alone (n = 3 replicates), and fibrinogen and thrombin with tPA loaded into nonbinding CS nanogels (n = 3 replicates) and tPA-C/S-IgG (n = 3 replicates). Alexa-Fluor 488–labeled fibrinogen was used in all fibrinogen solutions and the stationary clot. All nanogels used are fluorescently labeled (rhodamine B), and arrows point to nanogels present in the fluidic device above. Images were taken on an EVOS FL Auto Imaging System at 4× magnification, and the remaining conditions are shown in supplemental Figure 1. The solid line represents the initial clot boundary, and the dotted line represents the clot boundary after 20 minutes of flow at a wall shear rate of 1 s−1, which was used to quantify clot growth. (E) Quantification was performed using ImageJ software. Data are presented as average ± standard deviation. Clot dynamics were analyzed via a 1-way analysis of variance (ANOVA) with a Tukey’s post hoc test using a 95% confidence interval. Fib, fibrinogen; Th, thrombin. *P < .05; ***P < .001; ****P < .0001.
In vitro evaluation of drug release characteristics and clotting under static and dynamic conditions. (A) tPA loading efficacy comparing FSN and CS-IgG particles (n = 3 nanogel samples). (B) tPA release studies comparing FSN and CS-IgG particles (n = 3 nanogel samples). (C) Absorbance-based polymerization-degradation assay with tPA loaded particles (n = 3 clots per condition). (D) In vitro clotting under dynamic conditions in a fluidic device that contained a stationary fibrin clot located in the clot reservoir and that used flow solutions with fibrinogen and thrombin (control) (n = 7 replicates), fibrinogen and thrombin with unloaded FSNs (n = 4 replicates), fibrinogen and thrombin with nonbinding core-shell (CS) nanogels (n = 3 replicates), fibrinogen with unloaded FSNs without thrombin and thus no active coagulation (n = 3 replicates), fibrinogen and thrombin with tPA-FSNs (n = 4 replicates), fibrinogen and thrombin with tPA alone (n = 3 replicates), and fibrinogen and thrombin with tPA loaded into nonbinding CS nanogels (n = 3 replicates) and tPA-C/S-IgG (n = 3 replicates). Alexa-Fluor 488–labeled fibrinogen was used in all fibrinogen solutions and the stationary clot. All nanogels used are fluorescently labeled (rhodamine B), and arrows point to nanogels present in the fluidic device above. Images were taken on an EVOS FL Auto Imaging System at 4× magnification, and the remaining conditions are shown in supplemental Figure 1. The solid line represents the initial clot boundary, and the dotted line represents the clot boundary after 20 minutes of flow at a wall shear rate of 1 s−1, which was used to quantify clot growth. (E) Quantification was performed using ImageJ software. Data are presented as average ± standard deviation. Clot dynamics were analyzed via a 1-way analysis of variance (ANOVA) with a Tukey’s post hoc test using a 95% confidence interval. Fib, fibrinogen; Th, thrombin. *P < .05; ***P < .001; ****P < .0001.
Fibrin-specific drug-loaded nanogels release functional tPA and lyse clots in vitro
Next, the functionality of fibrin-specific tPA-loaded particles was tested. By using an absorbance-based polymerization and degradation assay that was conducted under static conditions, tPA-FSNs exhibited a dual functionality in that they promoted both fibrin polymerization and degradation. tPA-FSN fluctuating absorbance values showed fluctuating coagulations, indicating a modulation of polymerization and degradation not seen in controls. Importantly, the degradation characteristics observed through decreases in absorbance values at 20 and 40 minutes validate the release of functionally active tPA (Figure 2C).
We next investigated the procoagulant and fibrinolytic action of tPA-FSN to preexisting clots under conditions of flow that support fibrin polymerization to mimic DIC, particularly the prothrombotic disease state in which thrombin generation overpowers natural anticoagulant/fibrinolytic pathways. End point images and quantification of the stationary boundary clot growth show that under these dynamic conditions, clot growth in the controls occurs at the boundary and minimal polymerization is observed in upstream channels (Figure 2D-E; supplemental Figure 3). In the presence of unloaded FSNs, significant clot augmentation was observed. This was expected because unloaded FSNs target the clotting process by crosslinking fibrin fibers, and they assist in maturation of actively polymerizing clots.39 Supplemental Figure 4 shows enhanced fiber density in the clot microstructure upon the addition of FSNs compared with control clot and clots with CS-IgG particles. C/S nanogels did not significantly alter clotting dynamics compared with controls, and without active polymerization (+FSN and –Th), the effect FSNs have on clot augmentation is diminished. In the presence of tPA-FSNs in actively polymerizing conditions, little upstream clotting and significantly less clot augmentation at the boundary is observed compared with controls because tPA-FSNs promote fibrinolytic action. This effect on clot lysis is more prominent than tPA alone or with nontargeted tPA-C/S particles and tPA-CS-IgG particles. Overall, these studies conducted under flow suggest that tPA-FSNs can attenuate growing aberrant clot formations.
Microthrombi presentation is diminished and consumption of clotting factors is recovered in a rodent DIC model with tPA-FSN treatment
We next evaluated the ability of tPA-FSNs to modulate outcomes in vivo in an LPS-DIC model. DIC animals exhibited darkened organ color compared with control animals. MSB-stained tissue sections also indicate microthrombi formations in DIC organs compared with controls, which is not as visible in DIC animals treated with tPA-FSN. With the quantified fibrin IHC, significantly more microthrombi was seen in the heart, lungs, liver, and kidneys in DIC animals treated with saline or FSNs compared with controls (Figure 3A-E). However, significantly fewer microthrombi were seen in the heart, lungs, kidneys, and liver in animals treated with tPA-FSN, indicating fibrinolytic action from tPA-FSN treatment. Fewer microthrombi were also seen in DIC animals treated with tPA-CS-IgG in the liver and lungs. Organ weights are shown in supplemental Figure 4, and heart, lungs, and spleen showed no differences. A significant increase in kidney weight in DIC animals treated with saline, FSNs, and tPA-FSNs was seen compared with controls. A significant increase in liver weight in DIC animals treated with saline, FSNs, and tPA-FSNs was also seen compared with DIC plus tPA-CS-IgG particles.
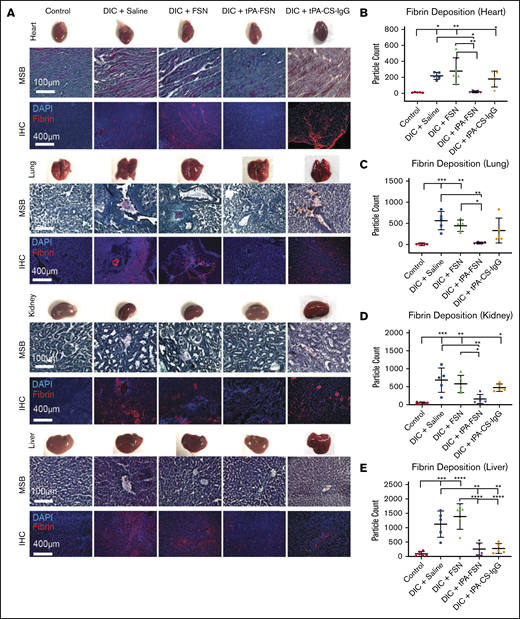
In vivo microthrombi examination in a DIC rodent model. (A) Shown are LPS-induced DIC rodent model harvested organs, including heart, lungs, kidneys, and liver, with corresponding photos of whole organs, MSB-stained tissue sections, and IHC for fibrin deposition in tissue sections. (B-E) DIC animals were treated with saline (n = 5), unloaded FSNs (n = 5), tPA-FSNs (n = 5), and tPA-CS-IgG (n = 5) and compared with controls (n = 6). Images were taken on an EVOS FL Auto Imaging System at 10× magnification. Corresponding quantification (performed with ImageJ particle analysis software) for IHC of fibrin deposition in heart (B), lung (C), kidney (D), and liver (E) tissue sections was averaged using 3 images per section for each animal (n = 6 for control liver, n = 4 for DIC + tPA-FSN lung, and n = 5 for all other groups). Data are presented as average ± standard deviation. Data sets were analyzed via 1-way ANOVA with a Tukey’s post hoc test using a 95% confidence interval. DAPI, 4′,6-diamidino-2-phenylindole. *P < .05; **P < .01; ***P < .001; ****P < .0001.
In vivo microthrombi examination in a DIC rodent model. (A) Shown are LPS-induced DIC rodent model harvested organs, including heart, lungs, kidneys, and liver, with corresponding photos of whole organs, MSB-stained tissue sections, and IHC for fibrin deposition in tissue sections. (B-E) DIC animals were treated with saline (n = 5), unloaded FSNs (n = 5), tPA-FSNs (n = 5), and tPA-CS-IgG (n = 5) and compared with controls (n = 6). Images were taken on an EVOS FL Auto Imaging System at 10× magnification. Corresponding quantification (performed with ImageJ particle analysis software) for IHC of fibrin deposition in heart (B), lung (C), kidney (D), and liver (E) tissue sections was averaged using 3 images per section for each animal (n = 6 for control liver, n = 4 for DIC + tPA-FSN lung, and n = 5 for all other groups). Data are presented as average ± standard deviation. Data sets were analyzed via 1-way ANOVA with a Tukey’s post hoc test using a 95% confidence interval. DAPI, 4′,6-diamidino-2-phenylindole. *P < .05; **P < .01; ***P < .001; ****P < .0001.
When examining coagulation parameters, an increase was observed in D-dimer in DIC animals compared with controls (Figure 4E). No significant differences were seen in fibrinogen levels upon induction of DIC or after treatment with either saline, FSNs, or tPA-FSNs (Figure 4F). The elevated fibrin degradation product, and somewhat reduced fibrinogen levels, helps validate fibrin turnover and microthrombi formation in this model. Decreased platelet count is another hallmark of DIC, and upon LPS-induced DIC, animals treated with saline, FSNs, or tPA-CS-IgGs showed significantly lower platelet counts compared with controls, indicating that FSNs or tPA alone had little effect on circulating platelets. However, after treatment with tPA-FSNs, platelet count recovered to control levels, indicating a recovery of the consumption of clotting factors known to occur in DIC (Figure 5G).
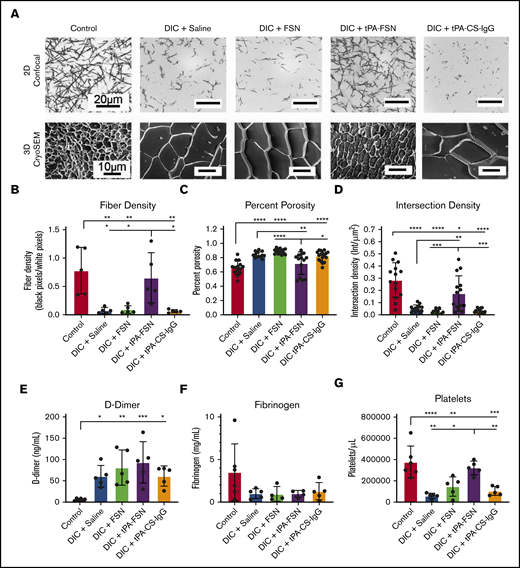
Examination of clotting characteristics and coagulation parameters from a DIC rodent model. (A) Clot formation from control and DIC animals treated with saline, FSNs, tPA-FSNs, or tPA-CS-IgG particles photographed using confocal microscopy (top) and cryoSEM (bottom). A Zeiss Laser Scanning Microscope (LSM 710, Zeiss Inc., White Plains, NY) was used for confocal images. A C-Apochromat 1.2W 63× objective lens was used for 1.89-μm z-stack images, which were analyzed by using ImageJ for making 8-bit 3D projections. (B) Fiber density was quantified by determining the ratio of black (fiber) over white (background) pixels in each binary image (average of 3 images per animal) (n = 5 for all groups). Clot structure was also assessed by using cryoSEM (JEOL 7600F) with a Gatan Alto Cryo-transfer system. Magnification 2500× for all images. (C-D) Quantification was performed by using a DiameterJ plugin for ImageJ software to measure percent porosity (C) and intersection density (D) using cryoSEM images (2 or 3 images per animal; n = 6 control animals; 5 animals for all DIC groups). D-dimer (E) and fibrinogen (F) from plasma samples taken from control animals (n = 6) and DIC animals (n = 4 for DIC + FSN and DIC + tPA-FSN group fibrinogen levels; n = 5 for all other groups). (G) Platelet count was measured for control animals (n = 6) and DIC animals (n = 5 for all groups). All data are presented as average ± standard deviation. Data sets were analyzed via a 1-way ANOVA with a Tukey’s post hoc test using a 95% confidence interval. *P < .05; **P < .01; ***P < .001; ****P < .0001.
Examination of clotting characteristics and coagulation parameters from a DIC rodent model. (A) Clot formation from control and DIC animals treated with saline, FSNs, tPA-FSNs, or tPA-CS-IgG particles photographed using confocal microscopy (top) and cryoSEM (bottom). A Zeiss Laser Scanning Microscope (LSM 710, Zeiss Inc., White Plains, NY) was used for confocal images. A C-Apochromat 1.2W 63× objective lens was used for 1.89-μm z-stack images, which were analyzed by using ImageJ for making 8-bit 3D projections. (B) Fiber density was quantified by determining the ratio of black (fiber) over white (background) pixels in each binary image (average of 3 images per animal) (n = 5 for all groups). Clot structure was also assessed by using cryoSEM (JEOL 7600F) with a Gatan Alto Cryo-transfer system. Magnification 2500× for all images. (C-D) Quantification was performed by using a DiameterJ plugin for ImageJ software to measure percent porosity (C) and intersection density (D) using cryoSEM images (2 or 3 images per animal; n = 6 control animals; 5 animals for all DIC groups). D-dimer (E) and fibrinogen (F) from plasma samples taken from control animals (n = 6) and DIC animals (n = 4 for DIC + FSN and DIC + tPA-FSN group fibrinogen levels; n = 5 for all other groups). (G) Platelet count was measured for control animals (n = 6) and DIC animals (n = 5 for all groups). All data are presented as average ± standard deviation. Data sets were analyzed via a 1-way ANOVA with a Tukey’s post hoc test using a 95% confidence interval. *P < .05; **P < .01; ***P < .001; ****P < .0001.
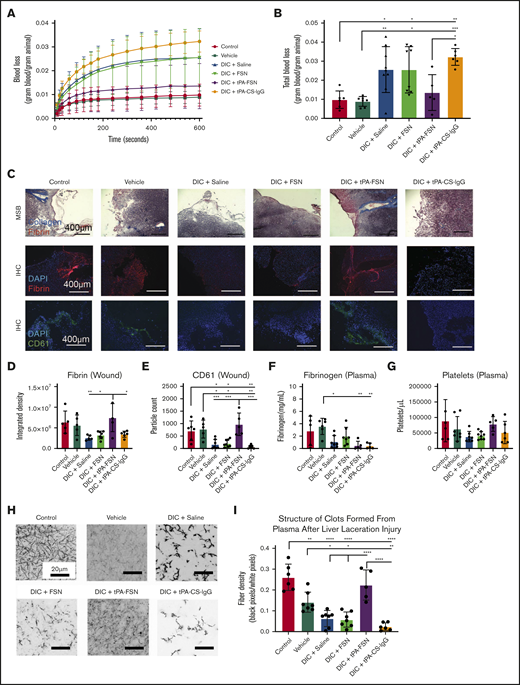
Evaluation of bleeding in a DIC rodent model with tPA-FSN treatment. Blood loss over time (A) and total blood loss (B) from a liver laceration injury from control (n = 6), vehicle (n = 7), and DIC animals treated with saline (n = 9), FSNs (n = 8), tPA-FSNs (n = 5), or tPA-CS-IgG (n = 6). (C) Representative histology images from wound tissue sections from each treatment group, including MSB-stained wound sections, IHC for fibrin at the wound site, and IHC for CD61 at the wound site. Images were taken on an EVOS FL Auto Imaging System at 10× magnification. Corresponding quantification was conducted with ImageJ particle analysis software. (D-E) IHC quantification at the wound sites for fibrin (D) (control, n = 5; vehicle, n = 5; DIC + saline, n = 5; DIC + FSN, n = 7; DIC + tPA-FSN, n = 5; DIC + tPA-CS-IgG, n = 6) and platelet CD61 (E) (control, n = 6; vehicle, n = 5; DIC + saline, n = 7; DIC + FSN, n = 8; DIC + tPA-FSN, n = 5; DIC + tPA-CS-IgG, n = 6). Blood samples from control, vehicle, and all DIC groups were used to determine fibrinogen levels (F) (control, n = 5; vehicle, n = 6; DIC + saline, n = 7; DIC + FSN, n = 7; DIC + tPA-FSN, n = 5; DIC + tPA-CS-IgG, n = 5) and platelet count (G) (control, n = 6; vehicle, n = 7; DIC + saline, n = 7; DIC + FSN, n = 8; DIC + tPA-FSN, n = 5; DIC + tPA-CS-IgG, n = 6). (H) Confocal microscopy was used to photograph clot formation for all treatment groups. (I) Corresponding quantification of fiber density (control, n = 6; vehicle, n = 7; DIC + saline, n = 7; DIC + FSN, n = 7; DIC + tPA-FSN, n = 5; DIC + tPA-CS-IgG, n = 6) with an average of 3 images per animal. A Zeiss LSM 710 Laser Scanning Microscope was used for confocal images. A C-Apochromat 63× 1.2W objective lens was used to capture 1.89-μm z-stack images, which were analyzed by using ImageJ to make 8-bit 3D projections. Fiber density was quantified by determining the ratio of black (fiber) over white (background) pixels in each binary image. Data are presented as average ± standard deviation. Data sets were analyzed via 1-way ANOVA with a Tukey’s post hoc test using a 95% confidence interval. *P < .05; **P < .01; ***P < .001; ****P < .0001.
Evaluation of bleeding in a DIC rodent model with tPA-FSN treatment. Blood loss over time (A) and total blood loss (B) from a liver laceration injury from control (n = 6), vehicle (n = 7), and DIC animals treated with saline (n = 9), FSNs (n = 8), tPA-FSNs (n = 5), or tPA-CS-IgG (n = 6). (C) Representative histology images from wound tissue sections from each treatment group, including MSB-stained wound sections, IHC for fibrin at the wound site, and IHC for CD61 at the wound site. Images were taken on an EVOS FL Auto Imaging System at 10× magnification. Corresponding quantification was conducted with ImageJ particle analysis software. (D-E) IHC quantification at the wound sites for fibrin (D) (control, n = 5; vehicle, n = 5; DIC + saline, n = 5; DIC + FSN, n = 7; DIC + tPA-FSN, n = 5; DIC + tPA-CS-IgG, n = 6) and platelet CD61 (E) (control, n = 6; vehicle, n = 5; DIC + saline, n = 7; DIC + FSN, n = 8; DIC + tPA-FSN, n = 5; DIC + tPA-CS-IgG, n = 6). Blood samples from control, vehicle, and all DIC groups were used to determine fibrinogen levels (F) (control, n = 5; vehicle, n = 6; DIC + saline, n = 7; DIC + FSN, n = 7; DIC + tPA-FSN, n = 5; DIC + tPA-CS-IgG, n = 5) and platelet count (G) (control, n = 6; vehicle, n = 7; DIC + saline, n = 7; DIC + FSN, n = 8; DIC + tPA-FSN, n = 5; DIC + tPA-CS-IgG, n = 6). (H) Confocal microscopy was used to photograph clot formation for all treatment groups. (I) Corresponding quantification of fiber density (control, n = 6; vehicle, n = 7; DIC + saline, n = 7; DIC + FSN, n = 7; DIC + tPA-FSN, n = 5; DIC + tPA-CS-IgG, n = 6) with an average of 3 images per animal. A Zeiss LSM 710 Laser Scanning Microscope was used for confocal images. A C-Apochromat 63× 1.2W objective lens was used to capture 1.89-μm z-stack images, which were analyzed by using ImageJ to make 8-bit 3D projections. Fiber density was quantified by determining the ratio of black (fiber) over white (background) pixels in each binary image. Data are presented as average ± standard deviation. Data sets were analyzed via 1-way ANOVA with a Tukey’s post hoc test using a 95% confidence interval. *P < .05; **P < .01; ***P < .001; ****P < .0001.
When evaluating the fibrin networks of clots formed from isolated plasma taken at the treatment end points, control animals formed robust clot networks with significantly higher fiber densities compared with DIC animals (Figure 4A-B), illustrating consumptive coagulopathy. DIC animals treated with FSN and tPA-CS-IgG did not recover clotting structure. However, an increase in fiber density was observed with tPA-FSN treatment, suggesting recovery of clotting factor consumption in these DIC animals. A similar trend was seen when ex vivo clots were analyzed using cryoSEM (Figure 4A; supplemental Figure 5). Specifically, clots formed from plasma from DIC animals treated with saline, FSN, or tPA-CS-IgG had a significantly higher percent porosity and lower intersection density compared with controls (Figure 4C-D), which was recovered in DIC animals treated with tPA-FSNs. Dot blots conducted with the plasma samples from control and DIC animals showed little presence of residual particles (supplemental Figure 6). In addition, exogenous addition of FSNs or tPA-FSNs to PPP from DIC plus saline PPP showed no improvement of clot structure, suggesting that improvements in the clot structure are due to the influence of the particles on consumptive coagulopathy while in circulation and are not due to residual particles in the plasma. Clotting characteristics of plasma samples from control and DIC animals shows that the consumption of clotting factors in DIC was apparent in this model, because DIC animals formed very poor clots ex vivo. However, clot structure was restored in animals receiving tPA-FSNs, which demonstrates the therapeutic efficacy of tPA-FSNs in this model. The biodistribution of these particles (supplemental Figure 7) observed after 30 minutes, showed a substantial presence of tPA-FSNs and FSNs in the kidneys. In addition, tPA-FSNs and FSN particles were detected in the lungs. To some extent, FSNs were detected in the heart and liver, and tPA-FSNs were detected in the spleen. Dot blot analysis shows significantly higher tPA-FSNs in whole blood samples compared with FSNs, and tPA-FSNs were present in the urine.
tPA-FSN treatment improved bleeding outcomes in the DIC rodent model
To directly evaluate the influence of tPA-FSN on bleeding risks associated with DIC, the LPS-DIC model was used in conjunction with a well-established liver injury bleeding model. Compared with control and vehicle groups, DIC-induced animals treated with saline, FSNs, or tPA-CS-IgGs bled more over time and had significantly higher total blood loss (Figure 5A-B). tPA-FSN treatment significantly decreased bleeding compared with tPA-CS-IgG, and there was no longer a significantly higher blood loss compared with control or vehicle groups (Figure 5A- B). MSB staining at the wound showed robust collagen and fibrin incorporation in tPA-FSN treatment groups (Figure 5C). IHC fibrin quantification showed significantly higher fibrin incorporation at the wound in DIC animals treated with tPA-FSNs compared with other DIC animals (Figure 5D). CD61 quantification showed that DIC animals treated with saline, FSNs, or tPA-CS-IgGs had significantly fewer platelets at the wound site compared with control, vehicle, or DIC animals treated with tPA-FSN (Figure 5E). When coupled with this injury model, fibrinogen levels in DIC animals treated with saline, tPA-FSN, or tPA-CS-IgG were significantly lower than in the vehicle group (Figure 5F), suggesting ongoing DIC or fibrinogen consumption at the wound sites. No significant differences were observed in platelet counts (Figure 5G). In addition, clotting capability in plasma samples after injury was significantly reduced in the DIC animals treated with saline, FSNs, or tPA-CS-IgGs and in the vehicle group compared with control animals. However, DIC animals treated with tPA-FSN had robust clotting capabilities after injury with significantly higher fiber density compared with other DIC animals (Figure 5H-I). Taken together, these data further demonstrate the ability of tPA-FSNs to improve bleeding outcomes in a DIC model potentially by ameliorating consumptive coagulopathy.
DIC patient plasma clot structure
To translate these therapeutic effects to human patients, nanogels were added to 3 DIC patient plasma samples ex vivo to evaluate clot structure. In these DIC patient samples, the average fibrinogen level was 335 ± 256 mg/dL, D-dimer level was 79 036 ± 29 648 ng/mL, prothrombin time was 23.7 ± 3.2 seconds, prothrombin international normalized ratio was 2.0 ± 0.3, activated partial thromboplastin time was 48.0 ± 23.3 seconds, and platelet count was 53 500 ± 9,192 platelets per μL. Individual values are shown in supplemental Table 1. Fibrin network properties of DIC patient plasma samples showed heterogeneous clot structure with significantly higher fiber densities when compared with controls (Figure 6A-B). Unloaded FSNs did not alter clot structure or fiber density; however, the addition of tPA-FSNs resulted in more homogeneous clot structures with reduced fiber density that more closely resembled clots from controls. The addition of tPA-CS-IgG particles resulted in areas of clot lysis mixed with highly heterogeneous areas of dense clot formation, which resulted in highly variable measurements of fiber density. Therefore, in this DIC patient population, tPA-FSN treatment could potentially be used to recover clotting capabilities similar to those in healthy human plasma.
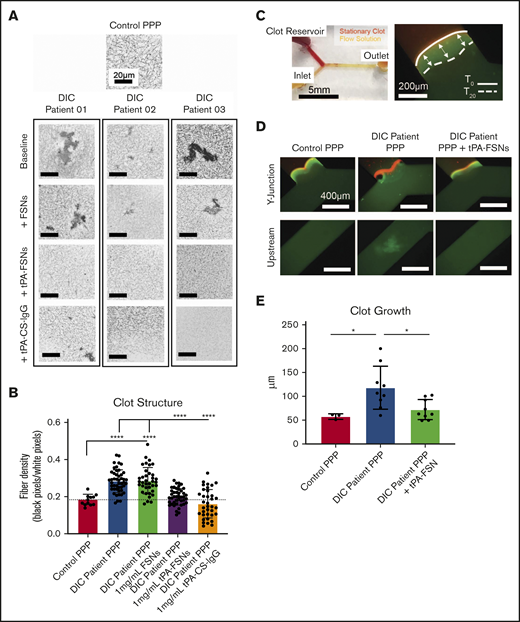
Effects of tPA-FSN treatment on ex vivo DIC patient plasma samples. Confocal microscopy (Zeiss LSM 710 Laser Scanning Microscope) images (A) and fiber density quantification (B) of DIC patient plasma clot samples compared with normal human plasma clots. A C-Apochromat 63× 1.2W objective lens was used to capture 1.89-μm z-stack images, which were analyzed using ImageJ to make 8-bit 3D projections. (B) Fiber density was quantified by determining the ratio of black (fiber) over white (background) pixels in each binary image. Unloaded FSNs, tPA-FSNs, or tPA-CS-IgG (1 mg/mL) were also incorporated into DIC patient plasma clots that were examined with confocal microscopy. Plasma samples from 3 DIC patients were examined, and pooled data from those samples was used for statistical analysis. (C) A custom-made Y-shaped microfluidic device was used to test plasma samples under flow at a wall shear rate of 10 s−1. A stationary fibrin clot was formed at the top Y junction with DIC patient plasma containing Alexa-Fluor 594–labeled fibrinogen for visualization. Flow solutions contained DIC patient plasma and Alexa-Fluor 488–labeled fibrinogen for visualization. (C-D) Images from an EVOS FL Auto Imaging System at 10× magnification were captured at the Y junction and upstream inlet channel and are shown with a reference to how clot growth was measured. (E) Quantification of clot growth at the stationary clot boundary was performed with ImageJ software. Normal healthy human plasma acted as a control. For each plasma sample from a DIC patient (n = 3), 3 trials were conducted with plasma alone in the flow solution, and 3 trials were conducted with plasma containing tPA-FSNs in the flow solution. All data are presented as average ± standard deviation. Data sets were analyzed via a 1-way ANOVA with a Tukey’s post hoc test using a 95% confidence interval. *P < .05; ****P < .0001.
Effects of tPA-FSN treatment on ex vivo DIC patient plasma samples. Confocal microscopy (Zeiss LSM 710 Laser Scanning Microscope) images (A) and fiber density quantification (B) of DIC patient plasma clot samples compared with normal human plasma clots. A C-Apochromat 63× 1.2W objective lens was used to capture 1.89-μm z-stack images, which were analyzed using ImageJ to make 8-bit 3D projections. (B) Fiber density was quantified by determining the ratio of black (fiber) over white (background) pixels in each binary image. Unloaded FSNs, tPA-FSNs, or tPA-CS-IgG (1 mg/mL) were also incorporated into DIC patient plasma clots that were examined with confocal microscopy. Plasma samples from 3 DIC patients were examined, and pooled data from those samples was used for statistical analysis. (C) A custom-made Y-shaped microfluidic device was used to test plasma samples under flow at a wall shear rate of 10 s−1. A stationary fibrin clot was formed at the top Y junction with DIC patient plasma containing Alexa-Fluor 594–labeled fibrinogen for visualization. Flow solutions contained DIC patient plasma and Alexa-Fluor 488–labeled fibrinogen for visualization. (C-D) Images from an EVOS FL Auto Imaging System at 10× magnification were captured at the Y junction and upstream inlet channel and are shown with a reference to how clot growth was measured. (E) Quantification of clot growth at the stationary clot boundary was performed with ImageJ software. Normal healthy human plasma acted as a control. For each plasma sample from a DIC patient (n = 3), 3 trials were conducted with plasma alone in the flow solution, and 3 trials were conducted with plasma containing tPA-FSNs in the flow solution. All data are presented as average ± standard deviation. Data sets were analyzed via a 1-way ANOVA with a Tukey’s post hoc test using a 95% confidence interval. *P < .05; ****P < .0001.
Evaluation of clotting dynamics of DIC patient plasma in a microdevice under active coagulation
After examining static conditions, DIC patient plasma was then incorporated into a microfluidic device to observe tPA-FSN efficacy under dynamic conditions. Under flow, baseline DIC patient plasma samples showed significantly higher clot growth at the stationary clot boundary site compared with controls, which was significantly decreased upon the addition of tPA-FSNs (Figure 6E). During coagulation under flow, DIC patient plasma also exhibited thrombosis in upstream channels (Figure 6D), and in some instances, in situ thrombus formation blocked flow. This was not observed in control human plasma samples or in DIC samples with tPA-FSNs. Overall, these fluidic conditions with patient plasma confirm the extensive disease state of DIC in which thrombosis is a critical concern. tPA-FSN incorporation in these studies indicates a potential therapeutic to mitigate thrombosis risks in this patient population.
Discussion
Through these studies, fibrin-targeting nanogels have been used to release tPA as a therapeutic strategy to treat widespread clotting and fibrin deposition under DIC circumstances. The fibrin-targeting potential of FSNs has been shown to augment clotting dynamics in conditions of active polymerization, which itself can aid in fibrin polymerization in circumstances of bleeding risks. With tPA-FSNs, fibrin-directed fibrinolysis was shown to more effectively lyse clots compared with undirected fibrinolytic action in vitro. In vivo, tPA-FSNs reduced microthrombi presentation, improved clot structure, and improved bleeding outcomes in LPS-induced DIC to a higher degree than their untargeted counterpart. tPA treatment in untargeted particles, while effective at reducing microthrombi presentation in some organs, did not improve bleeding outcomes. A number of sepsis-induced DIC models could have been considered to varying levels of physiological relevance, but the LPS-induced model replicated the thrombotic and consumptive coagulopathy of DIC in rodents which tPA-FSNs were designed to target.36,40-42 LPS-induced models displayed suppressed fibrinolytic-type DIC, similar to DIC types seen in sepsis.36 However, LPS-induced DIC models did not mimic all of the complex physiological responses to sepsis in human patients. For example, the rapid transient increase in cytokines displayed in the LPS-induced DIC model differ from those in human sepsis.40-42 In future studies, a cecal ligation and puncture model could be included to further support tPA-FSN efficacy in sepsis-induced thrombosis. Along with the alternative animal model consideration for future studies, blinded observations and randomized treatment allocation will be necessary to confirm therapeutic efficacy. Limitations in this study include unconscious bias as a result of unblinded observations.
The data presented in this study support the premise that tPA-FSNs can inhibit both microthrombi formation in organs and bleeding risks associated with DIC. In circumstances of consumptive coagulopathy with reduced platelets and clotting factors, tPA-FSNs may hone to aberrant microthrombi formations, lysing the clot to restore clotting factors such as platelets and also restore consumptive coagulopathy (Figure 4A-D,G). In addition, tPA-FSNs may bind to and prevent the growth of actively forming thrombi seen in vitro in Figures 2D-E and 6D-E, deterring further consumption of clotting factors. Indeed, when presented with an injury, DIC animals treated with tPA-FSNs exhibited blood loss comparable to that in controls. The decrease in bleeding compared with that in other DIC animals treated with saline, unloaded FSNs, or untargeted tPA delivery can be attributed to the action of the tPA-FSNs before the injury and their ability to recover consumptive coagulopathy. Systemic spillover of tPA is possible; however, with a nanogel delivery approach, potential spillover is limited to subtherapeutic doses in which increased bleeding was not observed. In human DIC patient samples, heterogeneous clot networks with areas of extremely high fiber densities demonstrated a prothrombotic disease state, perhaps before any consumption of clotting factors that can lead to bleeding complications. Under such circumstances, circulating tPA-FSNs can work to target fibrinolytic action in areas in which high fiber density or high fibrin content can cause disruptive microthrombi formation when in vivo. By mitigating these risks of aberrant clot formation with targeted fibrinolytic action, tPA-FSNs can potentially reduce thrombosis risks in this patient population. The timing of this therapeutic during progression of DIC should be thoughtfully considered and may not be appropriate in cases of active bleeding.
Anticoagulant therapies specifically have been studied to treat DIC. For example, antithrombin III (ATIII), when used without concomitant heparin, showed reduced mortality rates at 90 days, although with concomitant heparin, mortality did not differ between ATIII and placebo treatment.30 Bleeding incidences also increased with ATIII treatment, which is a crucial concern for DIC.30 Reducing the dosage of ATIII showed some promising results in improved 28-day mortality.43 By administering a more targeted fibrinolytic therapeutic, such as the tPA-FSNs examined in this study, bleeding risks would be attenuated because fibrinolytic action is localized to areas of fibrin accumulation and not off-target sites; thus, lower doses would inherently be required compared with systemic therapies. In addition, compared with ATIII therapies, tPA-FSNs would have the ability to eliminate preexisting aberrant microthrombi and not just new coagulation activity. Compared with heparin treatment, which in itself may be suitable only in certain DIC conditions, tPA-FSNs may offer reduced risks of off-target bleeding as well.14,44 Experimental treatments involving the thrombomodulin and protein C system, another strategy geared toward replenishing the native coagulation inhibitory mechanism in the coagulation pathway, have shown mixed results, with some studies reporting increased incidence of hemorrhage-related events, which again can be mitigated with a fibrin-targeted approach of tPA-FSNs.26,45-47 Important considerations for drug delivery systems used to treat thrombotic complications by delivering antithrombotic agents have been reviewed48 and studied for applications of thromboprophylaxis as well,49-52 which show promise in preventing thrombosis and providing protection against endotoxemia. tPA-FSNs may also act to direct therapeutic treatment by lysing existing aberrant thrombi and preventing further exacerbation of coagulation factors after initial onset of systemic coagulopathy.
This work has the potential to significantly improve treatment options for DIC patients. Although the coagulopathies associated with COVID-19 display distinct features compared with those of DIC, critically ill patients can progress to DIC as a result of additional secondary infections that are associated with higher mortality rates.53-55 Early work on administering anticoagulant treatment (eg, heparin) in COVID-19 patients with coagulopathy shows promise in reducing mortality, and these effects seem to be positively correlated with D-dimer levels.55 A case series of tPA treatment of COVID-19 patients with acute respiratory distress syndrome also showed initial improvements in lung function, although that improvement was not sustained after treatment with tPA.56 Because microcirculatory clot formation seems to be a prominent cause of acute lung injury and mortality in COVID-19 patients,57 tPA-FSNs may offer a novel therapeutic approach that could offer targeted delivery of fibrinolytic agents to lyse aberrant clots in the microcirculation to mitigate organ damage and potentially improve coagulopathy in this patient population.
These findings provide increased evidence for the need to develop targeted fibrinolytic strategies to treat thrombotic complications, including DIC, especially by targeting to preexisting fibrin. Next steps in this therapy should involve thorough safety analyses, including dosing studies. With a tunable nanogel platform that allows for multidrug encapsulation, dual-loaded studies with multiple therapeutics could be examined for enhanced therapeutic efficacy, especially those therapeutics that are directly related to mitigating the excessive thrombin generation that causes DIC. In conclusion, this study has shown that fibrin-targeting nanogels delivering tPA can help dissolve aberrant fibrin deposition to restore coagulation potential and thus mitigate thrombosis and bleeding complications of DIC, which will potentially improve outcomes.
For original data, please contact Ashley C. Brown at aecarso2@ncsu.edu.
Acknowledgments
The authors acknowledge E. Johannes and the Cellular and Molecular Imaging Facility for assistance with confocal microscopy and C. Zhou and the Analytical Instrumentation Facility at North Carolina State University.
This work was supported by grants from the National Science Foundation CAREER Division of Materials Research (1847488), the National Heart, Lung, and Blood Institute, National Institutes of Health (R01HL146701), a Flash grant from the North Carolina Biotechnology Center, an American Heart Association Predoctoral Fellowship (18PRE33990338) (E.P.M.), and CDMRP W81XWH-15-1-0485 from the US Department of Defense and National Institutes of Health, National Institute of General Medical Sciences T32 GM008600 (K.G.).
Authorship
Contribution: E.P.M. conducted particle synthesis and characterization, in vitro stationary and fluidic clotting dynamics experiments, in vivo studies with and without injury, subsequent analyses in these animal models, DIC patient sample microscopy, and fluidic device experiments and helped write the article; M.S. performed release studies comparing core-shell and single-layer particles, aided in particle synthesis and in vivo studies with the injury model, and helped write the article; N.M. conducted T-junction polydimethylsiloxane device fabrication, aided in T-junction device experiments, and helped write the article; K.N. helped with in vivo studies and conducted confocal microscopy in the DIC model without injury; K.G. and J.H.L. acquired DIC patient plasma; M.D. and H.D. supervised and fabricated microfluidic devices; and A.C.B. designed and supervised the study, analyzed the data, and helped write the article.
Conflict-of-interest disclosure: A.C.B. is a founder of SelSym Biotech, Inc., which is focused on developing injectable hemostatic materials. J.H.L. serves on advisory boards for Instrumentation Labs, Octapharma, and Merck. The remaining authors declare no competing financial interests.
Correspondence: Ashley C. Brown, Joint Department of Biomedical Engineering, North Carolina State University and University of North Carolina at Chapel Hill, 1001 William Moore Dr, Raleigh, NC 27606; e-mail: aecarso2@ncsu.edu

