

Key Points
In 9 unrelated families, we found 6 different TUBB1 variants, 1 of which novel, expanding the genetic spectrum of TUBB1-RT.
TUBB1 variants alter β1-tubulin localization and proplatelet formation, although they show high heterogeneity in clinical presentation.

Abstract
β1-Tubulin plays a major role in proplatelet formation and platelet shape maintenance, and pathogenic variants in TUBB1 lead to thrombocytopenia and platelet anisocytosis (TUBB1-RT). To date, the reported number of pedigrees with TUBB1-RT and of rare TUBB1 variants with experimental demonstration of pathogenicity is limited. Here, we report 9 unrelated families presenting with thrombocytopenia carrying 6 β1-tubulin variants, p.Cys12LeufsTer12, p.Thr107Pro, p.Gln423*, p.Arg359Trp, p.Gly109Glu, and p.Gly269Asp, the last of which novel. Segregation studies showed incomplete penetrance of these variants for platelet traits. Indeed, most carriers showed macrothrombocytopenia, some only increased platelet size, and a minority had no abnormalities. Moreover, only homozygous carriers of the p.Gly109Glu variant displayed macrothrombocytopenia, highlighting the importance of allele burden in the phenotypic expression of TUBB1-RT. The p.Arg359Trp, p.Gly269Asp, and p.Gly109Glu variants deranged β1-tubulin incorporation into the microtubular marginal ring in platelets but had a negligible effect on platelet activation, secretion, or spreading, suggesting that β1-tubulin is dispensable for these processes. Transfection of TUBB1 missense variants in CHO cells altered β1-tubulin incorporation into the microtubular network. In addition, TUBB1 variants markedly impaired proplatelet formation from peripheral blood CD34+ cell-derived megakaryocytes. Our study, using in vitro modeling, molecular characterization, and clinical investigations provides a deeper insight into the pathogenicity of rare TUBB1 variants. These novel data expand the genetic spectrum of TUBB1-RT and highlight a remarkable heterogeneity in its clinical presentation, indicating that allelic burden or combination with other genetic or environmental factors modulate the phenotypic impact of rare TUBB1 variants.
Introduction
The use of high-throughput sequencing (HTS) techniques1 has expanded the genetic spectrum of inherited thrombocytopenias (ITs), bringing the number of genes harboring variants responsible for IT up to around 40.2-7 Generally, ITs present with moderate thrombocytopenia and mild or no bleeding diathesis, although patients with severe thrombocytopenia can have clinically relevant hemorrhages.4,8,9 Some ITs associate with increased risk of developing severe disorders, including malignancies.2-5,8 Genetic variants in genes encoding for components of the actomyosin cytoskeleton or the microtubular system account for the most frequent forms of IT, which include myosin heavy chain‐IIA–related disorders (MYH9-RD), actinin 1–related thrombocytopenia (ACTN1-RT), and β1-tubulin–related thrombocytopenia (TUBB1-RT).3-5,10 Microtubules are assembled by heterodimers of α and β-tubulin, and β1-tubulin, encoded by TUBB1, is the predominant β-tubulin isoform in megakaryocytes (MKs) and platelets.11 Microtubules regulate proplatelet extension by MKs, and in platelets they form the marginal band, supporting their discoid shape.11-13 β1-tubulin knockout mice display impaired proplatelet formation and macrothrombocytopenia.14
We and others have reported common variants in β1-tubulin (p.Gln43Pro,15,16 p.Thr274Met,17 and p.Arg307His18 ). In particular, p.Gln43Pro is a frequent variant in patients with macrothrombocytopenia, which has been associated with protection from cardiovascular disease15 and increased risk of intracerebral hemorrhage.19 The p.Arg307His and p.Thr274Met variants have been related to lower platelet counts in Bernard-Soulier syndrome18 and immunethrombocytopenia20 and with less severe thrombocytopenia under paclitaxel treatment.17 These data suggest that common β1-tubulin variants can modulate platelet traits when combined with other genetic defects or acquired interfering factors.
A few rare TUBB1 variants (minor allele frequency < 1%) have been associated with thrombocytopenia in large case series of inherited platelet disorders (IPDs) investigated by HTS, but in most cases, experimental proof of their pathogenicity is still incomplete.21-27
Here, we characterize the largest series of TUBB1-RT cases reported thus far encompassing 38 individuals from 9 unrelated Spanish families bearing 6 different TUBB1 variants, including 1 novel missense variant. To appropriately assess the pathogenic effects of these variants, segregation and functional studies in patient platelets and mechanistic studies in cell models were performed.
Materials and methods
Patients, blood sampling, and DNA isolation
Patients with suspected IT and their relatives were recruited in the Spanish multicenter project “Functional and Molecular Characterization of Patients with Inherited Platelet Disorders.” The project obtained approval from the Ethics Committee of the Hospital Reina Sofía (Murcia, Spain) and follows the Helsinki Declaration rules. All participants gave written informed consent. Clinical data were reviewed, and bleeding symptoms were scored using the International Society on Thrombosis and Haemostasis bleeding assessment tool (ISTH-BAT).28,29 Venous blood samples were drawn into EDTA and sodium citrate for the different studies (complete blood count [CBC], immature platelet fraction [%IPF], blood smear and immunofluorescence, functional studies, electron microscopy, CD34+ cell isolation and MK cultures, DNA, and RNA).
Molecular analysis by HTS gene panel and Sanger sequencing
DNA from index cases was analyzed by extended HTS gene panels,24,30 designed with probes targeting all exons, 3′ untranslated region, and flanking regions of 89 genes related, or likely related, with IPDs (supplemental Table 1). The HTS process was carried out by using either a MiSeq Illumina platform (Illumina, San Diego, CA)24,30 or an Ion Torrent PGM platform (Thermo Fisher Scientific, Waltham, MA). Sequence data were mapped to Reference Human Genome (hg 19). Variant calling and annotation was performed using an in-house pipeline, based on VarScan v2.3.9, SAMTools v1.3.1, ANNOVAR, Ensembl-VEP v99, and dbNFSP v4.0a bioinformatic tools. Selected variants were confirmed and segregated in the families by Sanger sequencing. General information on the gene variants (chromosome position, Human Genome Variation Society [HGVS] name, Reference SNP cluster ID number [RS ID], frequency in different populations, pathogenicity and conservation scores, automated classification, ClinVar annotation, etc.) was initially obtained using the Varsome tool (https://varsome.com)31 (accessed May 2021). The guidelines of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology (ACMG/AMP),32 to assess the pathogenicity of the TUBB1 variants before and after taking into account the results of the current study, were followed. It should, however, be noted that, to date, specific ACMG/AMP rules for IPDs different from Glanzmann thrombasthenia33 and RUNX1-related disease34,35 have not been reported. Therefore, we adapted the ACMG criteria to the classification of TUBB1 variants of our patients as specified in supplemental Table 3.
Platelet tests
Platelet surface glycoprotein (GPs) expression, agonist-induced fibrinogen binding, and α- and δ-granule secretion (CD62 and CD63, respectively) were assessed by flow cytometry.30,36 Light transmission aggregometry was performed as previously described.37 Platelet adhesion and aggregation under high-shear stress conditions were also tested on the Impact-R cone-and-plate analyzer (Impact-R platelet analyzer 47600, Matis Medical Inc.), as described.38 Platelet morphology was analyzed by transmission electron microscopy.16 Immunofluorescence staining of tubulins and other cytoskeletal proteins was performed in washed platelets under resting and spreading conditions.39 Preplatelet and barbell proplatelet maturation was assessed in peripheral blood by immunofluorescence.40,41 Tubulin was detected in platelet lysates by immunoblotting.37,39 Platelet RNA was extracted using Trizol (ThermoFisher Scientific), and tubulin mRNA levels were measured using Taqman assays (ThermoFisher).39
MK cultures and proplatelet formation
Immunomagnetic isolation of CD34+ cells from peripheral blood samples, in vitro differentiation of MKs, immunofluorescence, and proplatelet formation studies were performed as previously reported.42,43
CHO cell models
The effect of the β1-tubulin missense variants was evaluated in CHO cells. For p.Arg359Trp, p.Thr107Pro, and p.Pher260Ser variants, the latter being a mutant previously reported as pathogenic, we used constructs consisting of N-myc tag fused to TUBB1 cDNA as previously described.22,23 For p.Gly269Asp and p.Gly109Glu variants, we used C-terminal DYKDDDDK (DYK) tag-fused TUBB1 cDNA constructs (GenScript, Leiden, The Netherlands). Cells were transiently transfected with either vector using Turbofect (ThermoFisher).
Additional methodologic details can be found in supplemental Methods.
Results
Patients
We investigated 38 patients and relatives from 9 unrelated Spanish families (Figure 1A). The index probands were referred for possible IPD because of lifelong macrothrombocytopenia although, for most of them, this was not associated with a relevant bleeding tendency (ISTH-BAT mean score ± standard deviation [SD]: 1.10 ± 2.07; median, 0; range, 0-8; Table 1). Thrombocytopenia, defined as platelet count below the normal healthy volunteer range (n = 107; 142-359 × 109/L), was in general moderate (median, 76; range, 57-134 × 109/L) and associated with increased platelet size (mean platelet volume [MPV], 13.3-15.0 fL; normal, 9.0-12.8 fL; Table 1). In several cases, MPV could not be reliably measured because of the abnormally wide platelet size distribution (supplemental Figure 1). Interestingly, we found that the %IPF, measured in some members of pedigrees F, H, and I, was abnormally high in thrombocytopenic patients (Table 1), especially in those carrying missense variants in β1-tubulin. Other blood parameters were normal (Table 1). In pedigree I, the parents (I.1 and I.2) of the probands (II.1 and II.2) were second cousins. In this family, several subjects across 3 generations displayed increased MPV, but only the 2 probands were thrombocytopenic (≈60 × 109/L at electronic counting), although they did not have excessive bleeding, including at childbirths and miscarriage (II.1). Microscopic platelet counting44 was ≈100 × 109/L in both siblings, with more than 20% of large platelets (mean platelet diameter, [MPD] > 5 µm) and occasional giant platelets (MPD > 6 µm; Figure 1B). In blood smears from the nonthrombocytopenic relatives, such as subject II.3 (Figure 1A), few (5%) large platelets (MPD > 5 µm) were present (Figure 1B; Table 1).
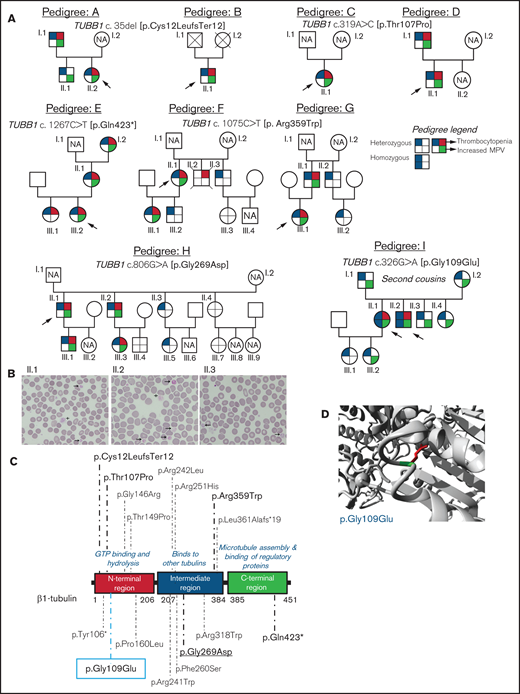
Family pedigrees and location of the β1-tubulin variants. (A) Pedigrees of the affected families with inherited thrombocytopenia. The index cases are indicated with black arrows. The upper right quarter red shading in symbols indicates individuals with thrombocytopenia, lower right quarter green shading denotes increased MPV, and upper and lower left quarter blue shading indicates heterozygous or homozygous for TUBB1 variant. NA, not available for the study; unlabeled symbols correspond to nonblood relatives not included in the study. (B) Representative peripheral blood smears from pedigree I after May-Grünwald Giemsa staining (×100). Variable platelet size was observed with large (arrows) and giant (cross) platelets. (C) Schematic representation of the β1-tubulin protein with all the reported variants. Variants in pedigrees reported in the present study are highlighted in bold; underlined variant is novel and the one within blue rectangle was found in homozygosity. (D) Structural analysis of the p.Gly109Glu missense variant using a β1-tubulin 3D model (software https://www3.cmbi.umcn.nl/hope/). The protein is colored in gray; the side chains of both the wild-type and the mutant residues are shown in green and red, respectively.
Family pedigrees and location of the β1-tubulin variants. (A) Pedigrees of the affected families with inherited thrombocytopenia. The index cases are indicated with black arrows. The upper right quarter red shading in symbols indicates individuals with thrombocytopenia, lower right quarter green shading denotes increased MPV, and upper and lower left quarter blue shading indicates heterozygous or homozygous for TUBB1 variant. NA, not available for the study; unlabeled symbols correspond to nonblood relatives not included in the study. (B) Representative peripheral blood smears from pedigree I after May-Grünwald Giemsa staining (×100). Variable platelet size was observed with large (arrows) and giant (cross) platelets. (C) Schematic representation of the β1-tubulin protein with all the reported variants. Variants in pedigrees reported in the present study are highlighted in bold; underlined variant is novel and the one within blue rectangle was found in homozygosity. (D) Structural analysis of the p.Gly109Glu missense variant using a β1-tubulin 3D model (software https://www3.cmbi.umcn.nl/hope/). The protein is colored in gray; the side chains of both the wild-type and the mutant residues are shown in green and red, respectively.
Blood parameters and clinical data of members of the reported families
| Pedigree | Case | Age, y | WBC × 109/L | RBC × 1012/L | Hb, g/dL | Hct (%) | Platelets × 109/L | MPV (fL) | ISTH-BAT | IPF (%) |
|---|---|---|---|---|---|---|---|---|---|---|
| A | I.1 | 75 | 6.57 | 4.97 | 14.3 | 42.5 | 84 | NR | 0 | ND |
| II.1 | 49 | 5.92 | 5.79 | 16.2 | 46.9 | 153 | 13.3 | 0 | ND | |
| II.2 | 43 | 6.08 | 4.56 | 13.7 | 39.5 | 124 | NR | 0 | ND | |
| B | II.1 | 74 | 7.4 | 3.82 | 11.1 | 34.3 | 65 | 13.4 | 2 | ND |
| C | II.1 | 21 | 5.19 | 4.64 | 14.2 | 41.1 | 70 | 14 | 2 | ND |
| D | I.1 | 54 | 5.9 | 4.78 | 14.1 | 43 | 110 | 14.5 | 1 | ND |
| II.1 | 14 | 3.05 | 3.78 | 11.6 | 35.5 | 85 | 15 | 2 | ND | |
| E | I.2 | 85 | 6 | 4.93 | 14.6 | 43.2 | 115 | 15 | 2 | ND |
| II.1 | 51 | 9.13 | 4.24 | 13 | 38.3 | 116 | 14.5 | 5 | ND | |
| III.1 | 21 | 7.6 | 4.94 | 14.2 | 42.2 | 131 | 12.4 | 3 | ND | |
| III.2 | 22 | 8.97 | 4.65 | 12.9 | 38.9 | 123 | 14.1 | 5 | ND | |
| F | II.1 | 65 | 5.45 | 4.74 | 13.5 | 41.2 | 134 | NR | 8 | 22.70 |
| II.2 | 65 | 10.15 | 4.87 | 15.7 | 47.5 | 100 | 12.4 | 0 | ND | |
| II.3 | 62 | 7.42 | 4.65 | 14.2 | 42.3 | 175 | 12.3 | 0 | ND | |
| III.1 | 31 | 7.19 | 4.68 | 13 | 39.2 | 118 | NR | 4 | 23.40 | |
| III.2 | 27 | 5.15 | 5.27 | 14.3 | 44.8 | 233 | 11.3 | 0 | 4.30 | |
| III.3 | 21 | 10.42 | 4.12 | 12.4 | 37.4 | 214 | 12.2 | 0 | 8.60 | |
| G | II.1 | 59 | 2.14 | 4.82 | 14.2 | 44.1 | 82 | 13.3 | 0 | ND |
| II.2 | 60 | 5.34 | 5.49 | 16 | 49.3 | 153 | 12.7 | 0 | ND | |
| III.1 | 36 | 5.74 | 4.89 | 15 | 46.9 | 69 | NR | 7 | ND | |
| III.2 | 4 | 5.4 | 5.21 | 16.3 | 51.8 | 261 | 10.7 | 0 | ND | |
| H | II.1 | 65 | 5.75 | 4.67 | 14.9 | 42.6 | 82 | NR | 0 | 24.00 |
| II.2 | 58 | 6.02 | 5.39 | 16.6 | 47.2 | 133 | 13.8 | 0 | 14.00 | |
| II.3 | 63 | 6.58 | 4.58 | 15.3 | 42.7 | 252 | 11.2 | 0 | 6.40 | |
| II.4 | 61 | 5.74 | 4.47 | 15.2 | 42.5 | 202 | 10.8 | 0 | 4.10 | |
| III.1 | 39 | 4.6 | 4.57 | 15.8 | 42.8 | 106 | 13.8 | 0 | ND | |
| III.3 | 24 | 6.28 | 4.72 | 14.5 | 41.6 | 104 | NR | 1 | ND | |
| III.4 | 27 | 5.78 | 5.34 | 16.5 | 46.6 | 202 | 11.8 | 0 | ND | |
| III.5 | 34 | 5.97 | 5.18 | 16.3 | 43.8 | 229 | 11.3 | 0 | ND | |
| III.7 | 22 | 6.33 | 4.89 | 15.2 | 43.2 | 264 | 10.2 | 0 | ND | |
| I | I.1 | 89 | 8.4 | 5.2 | 14.3 | 45 | 143 | 13.7 | 0 | ND |
| I.2 | 83 | 8 | 5.1 | 13.6 | 43.1 | 166 | NR | 0 | ND | |
| II.1 | 55 | 5.8 | 5.1 | 14.3 | 45 | 58 | NR | 0 | 37.30 | |
| II.2 | 51 | 10.6 | 7.1 | 16.9 | 53.3 | 57 | NR | 0 | 30.00 | |
| II.3 | 53 | 7.2 | 6.3 | 13.8 | 43.1 | 179 | 13.4 | 0 | 13.20 | |
| II.4 | 49 | 7.4 | 5.6 | 11.1 | 36 | 230 | NR | 0 | 10.30 | |
| III.1 | 19 | 4.7 | 4 | 11.6 | 35.8 | 205 | 12.4 | 0 | ND | |
| III.2 | 22 | 5.2 | 4.4 | 12.5 | 37.5 | 168 | 13.7 | 0 | ND |
| Pedigree | Case | Age, y | WBC × 109/L | RBC × 1012/L | Hb, g/dL | Hct (%) | Platelets × 109/L | MPV (fL) | ISTH-BAT | IPF (%) |
|---|---|---|---|---|---|---|---|---|---|---|
| A | I.1 | 75 | 6.57 | 4.97 | 14.3 | 42.5 | 84 | NR | 0 | ND |
| II.1 | 49 | 5.92 | 5.79 | 16.2 | 46.9 | 153 | 13.3 | 0 | ND | |
| II.2 | 43 | 6.08 | 4.56 | 13.7 | 39.5 | 124 | NR | 0 | ND | |
| B | II.1 | 74 | 7.4 | 3.82 | 11.1 | 34.3 | 65 | 13.4 | 2 | ND |
| C | II.1 | 21 | 5.19 | 4.64 | 14.2 | 41.1 | 70 | 14 | 2 | ND |
| D | I.1 | 54 | 5.9 | 4.78 | 14.1 | 43 | 110 | 14.5 | 1 | ND |
| II.1 | 14 | 3.05 | 3.78 | 11.6 | 35.5 | 85 | 15 | 2 | ND | |
| E | I.2 | 85 | 6 | 4.93 | 14.6 | 43.2 | 115 | 15 | 2 | ND |
| II.1 | 51 | 9.13 | 4.24 | 13 | 38.3 | 116 | 14.5 | 5 | ND | |
| III.1 | 21 | 7.6 | 4.94 | 14.2 | 42.2 | 131 | 12.4 | 3 | ND | |
| III.2 | 22 | 8.97 | 4.65 | 12.9 | 38.9 | 123 | 14.1 | 5 | ND | |
| F | II.1 | 65 | 5.45 | 4.74 | 13.5 | 41.2 | 134 | NR | 8 | 22.70 |
| II.2 | 65 | 10.15 | 4.87 | 15.7 | 47.5 | 100 | 12.4 | 0 | ND | |
| II.3 | 62 | 7.42 | 4.65 | 14.2 | 42.3 | 175 | 12.3 | 0 | ND | |
| III.1 | 31 | 7.19 | 4.68 | 13 | 39.2 | 118 | NR | 4 | 23.40 | |
| III.2 | 27 | 5.15 | 5.27 | 14.3 | 44.8 | 233 | 11.3 | 0 | 4.30 | |
| III.3 | 21 | 10.42 | 4.12 | 12.4 | 37.4 | 214 | 12.2 | 0 | 8.60 | |
| G | II.1 | 59 | 2.14 | 4.82 | 14.2 | 44.1 | 82 | 13.3 | 0 | ND |
| II.2 | 60 | 5.34 | 5.49 | 16 | 49.3 | 153 | 12.7 | 0 | ND | |
| III.1 | 36 | 5.74 | 4.89 | 15 | 46.9 | 69 | NR | 7 | ND | |
| III.2 | 4 | 5.4 | 5.21 | 16.3 | 51.8 | 261 | 10.7 | 0 | ND | |
| H | II.1 | 65 | 5.75 | 4.67 | 14.9 | 42.6 | 82 | NR | 0 | 24.00 |
| II.2 | 58 | 6.02 | 5.39 | 16.6 | 47.2 | 133 | 13.8 | 0 | 14.00 | |
| II.3 | 63 | 6.58 | 4.58 | 15.3 | 42.7 | 252 | 11.2 | 0 | 6.40 | |
| II.4 | 61 | 5.74 | 4.47 | 15.2 | 42.5 | 202 | 10.8 | 0 | 4.10 | |
| III.1 | 39 | 4.6 | 4.57 | 15.8 | 42.8 | 106 | 13.8 | 0 | ND | |
| III.3 | 24 | 6.28 | 4.72 | 14.5 | 41.6 | 104 | NR | 1 | ND | |
| III.4 | 27 | 5.78 | 5.34 | 16.5 | 46.6 | 202 | 11.8 | 0 | ND | |
| III.5 | 34 | 5.97 | 5.18 | 16.3 | 43.8 | 229 | 11.3 | 0 | ND | |
| III.7 | 22 | 6.33 | 4.89 | 15.2 | 43.2 | 264 | 10.2 | 0 | ND | |
| I | I.1 | 89 | 8.4 | 5.2 | 14.3 | 45 | 143 | 13.7 | 0 | ND |
| I.2 | 83 | 8 | 5.1 | 13.6 | 43.1 | 166 | NR | 0 | ND | |
| II.1 | 55 | 5.8 | 5.1 | 14.3 | 45 | 58 | NR | 0 | 37.30 | |
| II.2 | 51 | 10.6 | 7.1 | 16.9 | 53.3 | 57 | NR | 0 | 30.00 | |
| II.3 | 53 | 7.2 | 6.3 | 13.8 | 43.1 | 179 | 13.4 | 0 | 13.20 | |
| II.4 | 49 | 7.4 | 5.6 | 11.1 | 36 | 230 | NR | 0 | 10.30 | |
| III.1 | 19 | 4.7 | 4 | 11.6 | 35.8 | 205 | 12.4 | 0 | ND | |
| III.2 | 22 | 5.2 | 4.4 | 12.5 | 37.5 | 168 | 13.7 | 0 | ND |
Index cases in each pedigree are highlighted in bold. Underlined cases are non carriers of β1-tubulin variants. Normal range in healthy subjects from our cohort (n = 107): platelets, 142 to 359 × 109/L; and MPV, 9 to 12.8 fL. The IPF (%) in control subjects (n = 6), assayed in parallel with patients, ranged from 2.30 to 3.30. Hb, hemoglobin; Hct, hematocrit; ND, not determined; NR, not recorded; RBC, red blood cells; WBC, white blood cells.
Identification of rare variants in β1-tubulin by HTS
Analysis of DNA with the HTS gene panel identified 6 different candidate variants in TUBB1 (NM_030773.4). Sanger sequencing confirmed these variants in the probands and in several relatives (Figure 1A).
In pedigrees A and B, the frameshift c.35del [p.Cys12LeufsTer12]24 was found, whereas families C and D carried the missense variant c.319A>C [p.Thr107Pro].24 The nonsense variant c.1267C>T [p.Gln423*]24 was identified in family E, and the missense variant c.1075C>T [p. Arg359Trp]24 was present in pedigrees F and G. Family H carried the novel missense variant c.806G>A [p.Gly269Asp]. All these variants were found in heterozigosity and affected conserved residues in different domains of the β1-tubulin protein (Figure 1C). Noteworthy, the phenotypic effect of these variants varied among family members: in pedigrees A, E, F, G, and H, some carriers presented with macrothrombocytopenia, whereas others displayed only increased MPV and few normal platelet count and size (Figure 1A; Table 1). Thrombocytopenia in a noncarrier of TUBB1 variants (II.2, pedigree F; who died of sudden myocardial infarction) was attributed to habitual alcohol intake and thus interpreted as acquired.
An interesting finding was the identification in pedigree I of the c.326G>A [p.Gly109Glu] TUBB1 variant in exon 4 of TUBB1, leading to the p.Gly109Glu substitution that affects a highly conserved residue at the N-terminal domain of β1-tubulin (Figure 1C-D). The distinctive feature of this variant is that, although the homozygous probands II.1 and II.2 displayed macrothombocytopenia, the heterozygous relatives, except one, had only increased MPV.
The p.Thr107Pro, p.Gly109Glu, and p.Gly269Asp variants were predicted to be pathogenic by a higher number of softwares and to be 100% conserved during evolution compared with the p.Arg359Trp variant (supplemental Table 2). In silico modeling of protein structure predicted that all variants would disturb correct protein folding. In p.Gly109Glu and p.Gly269Asp a neutral charge residue is replaced by a negatively charged one; moreover, Gly is the most flexible of all the aminoacids and thus its substitution may lead to the loss of the correct torsion angles of the protein backbone with a possible stronger impact on protein structure. Concerning variants p.Thr107Pro and p.Arg359Trp, a more hydrophobic residue is introduced, and this may cause loss of hydrogen bonds and/or disturb correct protein folding (Figure 1D; supplemental Figure 2). According to the available evidence (population, computational, functional, and segregation data), and based on the ACMG criteria (adapted for the TUBB1 gene), all variants were initially classified as of uncertain significance (VUS), except c.1267C>T [p.Gln423*], which was classified as likely pathogenic (supplemental Table 3). However, this initial classification was modified based on the results of this study, and only c.1075C>T [p.Arg359Trp] and c.326G>A [p.Gly109Glu] remained as VUS (supplemental Table 3).
Assessment of the deleterious effect of missense TUBB1 variants on microtubule assembly
To assess the effect of the missense β1-tubulin variants on microtubule assembly, we expressed wild-type and mutant β1-tubulin in transfected CHO cells. As positive control, CHO cells were transfected with the mutant Ser260 β1-tubulin.23 As shown in Figure 2, wild-type β1-tubulin expressed in CHO cells readily incorporated into microtubules. In contrast, mutant Pro107 β1-tubulin showed an altered, diffuse pattern of cytoplasmic distribution, resembling Ser260. Trp359 β1-tubulin incorporated in part into microtubules but was also diffused in cytoplasm, and Asp269 and Glu109 mutants resulted in β1-tubulin, accumulating in punctiform aggregates in the cytoplasm (Figure 2). Moreover, microtubule length in CHO cells transfected with β1-tubulin mutants was significantly shorter compared with that in cells transfected with wild-type β1-tubulin (Figure 2C).
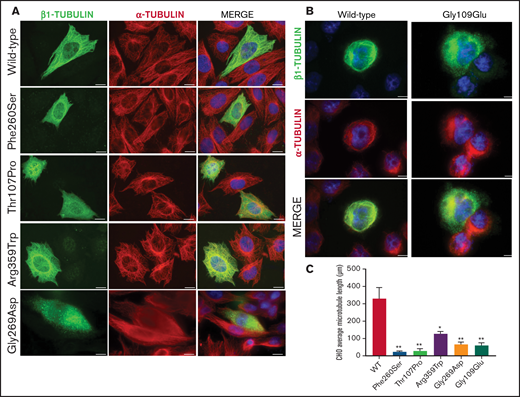
Effect of the novel missense variants in a CHO cellular model. Distribution of (A) transfected wild-type β1-tubulin, mutants p.260Ser, p.107Pro, p.359Trp, and p.269Asp and (B) mutant p.109Glu (in a different set of experiments), in transfected CHO cells by immunostaining for β1- (green) and α-tubulin (red). (C) Microtubule length in CHO cells transfected with wild-type or mutant β1-tubulin was assessed using ImageJ software. All β1-tubulin mutants resulted in significantly shorter microtubules. All images were acquired with a Carl Zeiss Axio Observer A1 fluorescence microscope with a 63× objective lens. Scale bars are 20 μm. Data are means ± SD of values obtained from at least 10 different microscopy fields. *P < .05; **P < .005.
Effect of the novel missense variants in a CHO cellular model. Distribution of (A) transfected wild-type β1-tubulin, mutants p.260Ser, p.107Pro, p.359Trp, and p.269Asp and (B) mutant p.109Glu (in a different set of experiments), in transfected CHO cells by immunostaining for β1- (green) and α-tubulin (red). (C) Microtubule length in CHO cells transfected with wild-type or mutant β1-tubulin was assessed using ImageJ software. All β1-tubulin mutants resulted in significantly shorter microtubules. All images were acquired with a Carl Zeiss Axio Observer A1 fluorescence microscope with a 63× objective lens. Scale bars are 20 μm. Data are means ± SD of values obtained from at least 10 different microscopy fields. *P < .05; **P < .005.
On the contrary, none of these mutants affected the assembly of α-tubulin into microtubules (Figure 2).
The observation of these deleterious effects prompted more extensive platelet phenotyping of patients, except for probands from pedigrees C and D, which were not available for these set of experiments.
β1-tubulin missense variants do not affect platelet function
Flow cytometry of platelets from p.Arg359Trp, p.Gly269Asp, and p.Gly109Glu carriers with thrombocytopenia showed levels of major GPs in the upper normal range (supplemental Figure 3A), compatible with increased platelet size. Agonist-induced fibrinogen binding, despite high variability, showed a trend to be increased in these individuals, and secretion of α- or δ-granules (CD62 and CD63 surface expression, respectively) was similar to that in control platelets (supplemental Figure 3B-D). Impact-R assay, where shear stress is applied to blood in a polystyrene well, showed no significant effect of variant p.Gly109Glu on platelet adhesion and aggregation (supplemental Figure 4). Moreover, homozygous and heterozygous carriers of p.Gly109Glu showed normal platelet aggregation response to different agonists (supplemental Figure 5).
Nonthrombocytopenic carriers of any of these 3 missense variants and noncarrier relatives showed normal platelet function tests (data not shown).
β1-tubulin missense variants alter β1-tubulin expression, platelet microtubule organization, spreading, proplatelet formation, and platelet maturation
Effect of the c.1075 C>T [p.Arg359Trp] variant.
In resting control platelets, β1-tubulin was localized in the microtubules of the marginal band. In contrast, in some platelets from carriers of the p.Arg359Trp variant (pedigree F), the β1-tubulin ring was slightly disorganized, suggesting a moderate detrimental effect of this variant on microtubule assembly. This effect was more evident in thrombocytopenic carriers (Figure 3A). β1-tubulin appeared disorganized also in spread platelets from variant carriers, lacking the normal distribution in fibers (Figure 3B). Despite this, the percentage of platelet spreading was normal (Figure 3C). Moreover, immunoblotting of platelet lysates showed that β1-tubulin expression was only minimally affected by the p.Arg359Trp variant (Figure 3D).
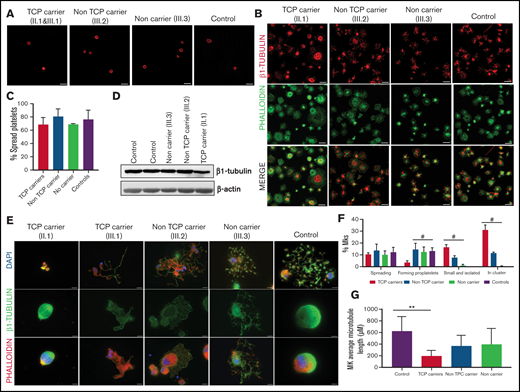
Effect of the p.Arg359Trp β1-tubulin variant in platelets and megakaryocytes. The functional and structural consequences of p.Arg359Trp β1-tubulin variant were assessed in members of Pedigree F. Immunofluorescence analysis of β1-tubulin was performed in platelets on poly-l-lysine–coated coverslips under (A) resting (platelet-rich plasma) and (B) spreading (washed platelets) conditions from p.Arg359Trp carriers with (II.1 and III.1) or without thrombocytopenia (III.2), a noncarrier (III.3), and an unrelated healthy control. Platelets were labeled with a β1-tubulin antibody (red) and with fluorescein isothiocyanate-phalloidin (green). Scale bars are 5 μm, and the objective lens is 63×. (C) Bar plot showing the percentage of spread platelets relative to total adhered platelets. Data are means ± SD of values obtained from at least 10 different microscopy fields in patient and control samples. (D) Representative Western blot picture of β1-tubulin levels in platelet lysates from p.Arg359Trp carriers with or without thrombocytopenia, a noncarrier, and 2 healthy controls, using β-actin as internal control. (E) Illustrative images of CD34+ peripheral blood cell–derived megakaryocytes in patients from Pedigree F (p.Arg359Trp carriers with or without thrombocytopenia and a noncarrier) and healthy controls. MKs were labeled with β1-tubulin (green) and rhodamine-phalloidin (red). Nuclei were stained with 4′,6-diamidino-2-phenylindole (DAPI; blue). Images were acquired in a Carl Zeiss Axio Observer A1 fluorescence microscope with a 63× objective lens. Scale bars are 5 μm. (F) Classification of MKs in family members and controls. Polynucleated cells extending protrusions with terminal tips were considered as proplatelet-forming MKs, whereas those displaying a flattened shape with actin organized in focal adhesion points and fibers as spread MKs. Some MKs were small and isolated, whereas MKs that were present in aggregates of 2 or more MKs were defined clusters. At least 100 MKs from 5 different replicates were analyzed; #P < .05 vs TCP carriers. (G) Microtubule length in CD34+ peripheral blood cell–derived MKs from controls and patients with the p.Arg359Trp β1-tubulin variant was assessed using the ImageJ software. Data are mean ± SD of values obtained from at least 6 different microscopy fields. TCP, thrombocytopenic; non-TCP, nonthrombocytopenic. **P < .005.
Effect of the p.Arg359Trp β1-tubulin variant in platelets and megakaryocytes. The functional and structural consequences of p.Arg359Trp β1-tubulin variant were assessed in members of Pedigree F. Immunofluorescence analysis of β1-tubulin was performed in platelets on poly-l-lysine–coated coverslips under (A) resting (platelet-rich plasma) and (B) spreading (washed platelets) conditions from p.Arg359Trp carriers with (II.1 and III.1) or without thrombocytopenia (III.2), a noncarrier (III.3), and an unrelated healthy control. Platelets were labeled with a β1-tubulin antibody (red) and with fluorescein isothiocyanate-phalloidin (green). Scale bars are 5 μm, and the objective lens is 63×. (C) Bar plot showing the percentage of spread platelets relative to total adhered platelets. Data are means ± SD of values obtained from at least 10 different microscopy fields in patient and control samples. (D) Representative Western blot picture of β1-tubulin levels in platelet lysates from p.Arg359Trp carriers with or without thrombocytopenia, a noncarrier, and 2 healthy controls, using β-actin as internal control. (E) Illustrative images of CD34+ peripheral blood cell–derived megakaryocytes in patients from Pedigree F (p.Arg359Trp carriers with or without thrombocytopenia and a noncarrier) and healthy controls. MKs were labeled with β1-tubulin (green) and rhodamine-phalloidin (red). Nuclei were stained with 4′,6-diamidino-2-phenylindole (DAPI; blue). Images were acquired in a Carl Zeiss Axio Observer A1 fluorescence microscope with a 63× objective lens. Scale bars are 5 μm. (F) Classification of MKs in family members and controls. Polynucleated cells extending protrusions with terminal tips were considered as proplatelet-forming MKs, whereas those displaying a flattened shape with actin organized in focal adhesion points and fibers as spread MKs. Some MKs were small and isolated, whereas MKs that were present in aggregates of 2 or more MKs were defined clusters. At least 100 MKs from 5 different replicates were analyzed; #P < .05 vs TCP carriers. (G) Microtubule length in CD34+ peripheral blood cell–derived MKs from controls and patients with the p.Arg359Trp β1-tubulin variant was assessed using the ImageJ software. Data are mean ± SD of values obtained from at least 6 different microscopy fields. TCP, thrombocytopenic; non-TCP, nonthrombocytopenic. **P < .005.
As shown in Figure 3E-F, proplatelet formation by CD34+ peripheral blood cell–derived MKs was almost absent in thrombocytopenic carriers of the p.Arg359Trp variant, with only few small proplatelets observed and mostly small and clustered MKs (Figure 3E). β1-tubulin organization was altered, as shown by decreased microtubule length (Figure 3G). In contrast, proplatelet formation was normal in nonthrombocytopenic carriers and noncarriers of this variant (Figure 3E-G). Finally, we observed a reduced number of circulating barbell-proplatelets and “figure 8” structures, and an increased number of preplatelets, showing an impaired final platelet maturation in blood (supplemental Figure 6A).
Effect of the c.806 G>A [p.Gly269Trp] variant.
In resting platelets from p.Gly269Trp variant carriers of pedigree H, either with or without thrombocytopenia, the β1-tubulin ring was missing, and tubulin was diffused in the cytoplasm. In contrast, noncarriers displayed the normal β1-tubulin marginal band (Figure 4A). Immunoblotting showed moderately reduced expression of β1-tubulin in platelets from p.Gly269Trp carriers (Figure 4B).
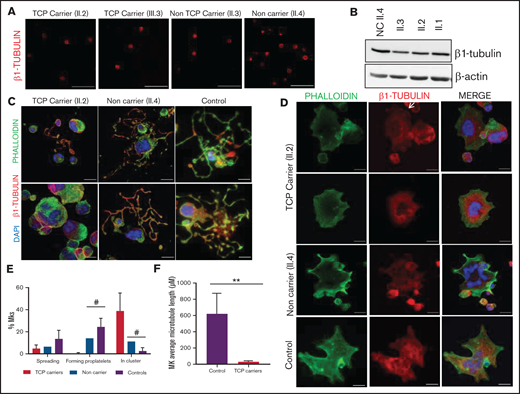
Effect of the p.Gly269Asp β1-tubulin variant in platelets and megakaryocytes. The functional and structural consequences of the novel p.Gly269Asp β1-tubulin variant were assessed in members of Pedigree H. (A) Immunofluorescence analysis of β1-tubulin was performed on poly-l-lysine–coated coverslips in platelets (platelet-rich plasma) from p.Gly269Asp carriers with (II.2 and III.3) or without thrombocytopenia (II.3) and a noncarrier (II.4). Platelets were labeled with a β1-tubulin antibody (red). Scale bars are 5 μm, and the objective lens is 63× (magnification 3×). (B) Western blot pictures of β1-tubulin levels in platelet lysates; β-actin was used as an internal control. (C) Representative images of CD34+ peripheral blood cell–derived MKs in a thrombocytopenic p.Gly269Asp carrier (II.2), noncarrier (II.4), and healthy control. (D) Representative images of spread MKs. MKs were labeled with β1-tubulin (green) and rhodamine-phalloidin (red). Nuclei were stained with DAPI (blue). The white arrow indicates a MKs with disorganized β1-tubulin. Images were acquired in a Leica SP8 confocal microscope with a 63× objective lens (magnification 1.5×). Scale bars are 5 μm. (E) Classification of MKs in family members and controls (as described in Figure 3; #P < .05 vs TCP carriers). (F) Microtubule length of CD34+ peripheral blood cell–derived MKs from controls and patients with p.Gly269Asp β1-tubulin variant was assessed using ImageJ software. Values are mean ± SD of values obtained from at least 10 different microscopy fields. TCP, thrombocytopenic; non-TCP, nonthrombocytopenic. **P < .005.
Effect of the p.Gly269Asp β1-tubulin variant in platelets and megakaryocytes. The functional and structural consequences of the novel p.Gly269Asp β1-tubulin variant were assessed in members of Pedigree H. (A) Immunofluorescence analysis of β1-tubulin was performed on poly-l-lysine–coated coverslips in platelets (platelet-rich plasma) from p.Gly269Asp carriers with (II.2 and III.3) or without thrombocytopenia (II.3) and a noncarrier (II.4). Platelets were labeled with a β1-tubulin antibody (red). Scale bars are 5 μm, and the objective lens is 63× (magnification 3×). (B) Western blot pictures of β1-tubulin levels in platelet lysates; β-actin was used as an internal control. (C) Representative images of CD34+ peripheral blood cell–derived MKs in a thrombocytopenic p.Gly269Asp carrier (II.2), noncarrier (II.4), and healthy control. (D) Representative images of spread MKs. MKs were labeled with β1-tubulin (green) and rhodamine-phalloidin (red). Nuclei were stained with DAPI (blue). The white arrow indicates a MKs with disorganized β1-tubulin. Images were acquired in a Leica SP8 confocal microscope with a 63× objective lens (magnification 1.5×). Scale bars are 5 μm. (E) Classification of MKs in family members and controls (as described in Figure 3; #P < .05 vs TCP carriers). (F) Microtubule length of CD34+ peripheral blood cell–derived MKs from controls and patients with p.Gly269Asp β1-tubulin variant was assessed using ImageJ software. Values are mean ± SD of values obtained from at least 10 different microscopy fields. TCP, thrombocytopenic; non-TCP, nonthrombocytopenic. **P < .005.
MKs from p.Gly269Trp carriers, differently from those of noncarriers and controls, were mainly clustered, MKs-forming proplatelets were almost absent with only some abnormally short proplatelets observed (Figure 4C,E). In addition, MKs from these patients adhered and spread normally on fibrinogen. β1-tubulin incorporated in microtubules in some MKs, whereas others showed an aberrant β1-tubulin distribution (Figure 4D) and shorter microtubules (Figure 4F), resembling the pattern observed in platelets and in the CHO cell model. We also observed reduced barbell-proplatelets and “figure 8” structures and increased preplatelets in blood, indicating an impaired final platelet maturation (supplemental Figure 6B).
Effect of the p.Gly109Glu variant.
Pedigree I was peculiar because only homozygous, and not heterozygous, carriers presented with macrothrombocytopenia.
Electron microscopy showed that most platelets from homozygous p.Gly109Glu carriers were round and large, with a slightly hyperthrophic open canalicular system and an irregular membrane demarcation system (Figure 5). In contrast, in heterozygous carriers the platelet population was heterogeneous in size and shape.
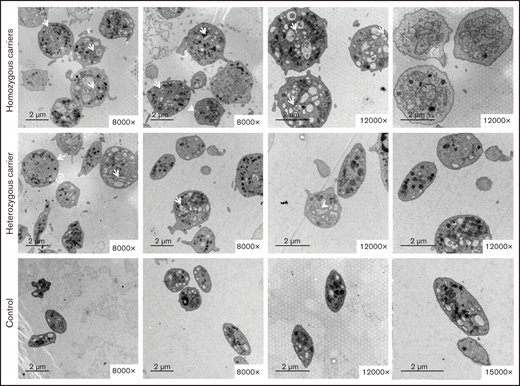
The variant Gly109Glu in β1-tubulin alters platelet ultrastructure. Electron microscopy of platelets from 2 homozygous and a heterozygous carrier of the β1-tubulin p.Gly109Glu and from 2 healthy controls. In contrast to control platelets, most platelets from homozygous p.Gly109Glu carriers were round and large and showed a slightly hypertrophic open canalicular system and an irregular membrane demarcation system (white arrows and asterisks, respectively). Magnification for each image is shown (8000-15 000×). Images were acquired in a Philips/FEITecnai12 transmission electron microscope.
The variant Gly109Glu in β1-tubulin alters platelet ultrastructure. Electron microscopy of platelets from 2 homozygous and a heterozygous carrier of the β1-tubulin p.Gly109Glu and from 2 healthy controls. In contrast to control platelets, most platelets from homozygous p.Gly109Glu carriers were round and large and showed a slightly hypertrophic open canalicular system and an irregular membrane demarcation system (white arrows and asterisks, respectively). Magnification for each image is shown (8000-15 000×). Images were acquired in a Philips/FEITecnai12 transmission electron microscope.
Platelet immunofluorescence confirmed that the detrimental effect of this variant on microtubule assembly is determined by allele burden. Resting platelets from p.Gly109Glu homozygous carriers lacked the marginal band, and β1-tubulin was disposed in aggregates in the cytoplasm, whereas the β1-tubulin marginal band was normal in platelets from heterozygous carriers (Figure 6A). Moreover, although platelets from homozygous siblings displayed normal spreading (Figure 6B), β1-tubulin formed aggregates localized in the cytoplasm instead than at the center of platelets, as observed in heterozygous carriers and controls (Figure 6C). In addition, β1-tubulin was undetectable in platelet lysates from homozygous carriers by immunoblotting using 2 different antibodies (Figure 6D). The cell distribution of other cytoskeletal proteins, such as actin, α-actinin, DIAPH1, filamin, and MYH9, was instead normal in these patients (supplemental Figure 7).

The p.Gly109Glu variant disrupts β1-tubulin incorporation into microtubules with a minimal effect on platelet spreading. Representative immunofluorescence analysis of β1-tubulin in patient (Pedigree I) and control washed platelets in (A) resting and (B-C) spreading conditions. Platelets were labeled with a β1-tubulin antibody (red) and with FITC-phalloidin (green). Images were acquired in a Carl Zeiss Axio Observer.A1 fluorescence microscope with a 100× objective lens. Scale bars are 5 μm. Platelet spreading on poly-l-lysine was quantified, and the percentage of totally spread platelets relative to the total adhered platelets is shown in the bar plot. Values are mean ± SD. Images were acquired in a Leica SP8 confocal microscope with a 63× objective lens (magnification, 2×). Scale bars are 20 μm. (D) Western blot pictures of β1-tubulin levels in platelet lysates from patients and controls, using 2 different β1-tubulin antibodies; β-actin was used as internal control.
The p.Gly109Glu variant disrupts β1-tubulin incorporation into microtubules with a minimal effect on platelet spreading. Representative immunofluorescence analysis of β1-tubulin in patient (Pedigree I) and control washed platelets in (A) resting and (B-C) spreading conditions. Platelets were labeled with a β1-tubulin antibody (red) and with FITC-phalloidin (green). Images were acquired in a Carl Zeiss Axio Observer.A1 fluorescence microscope with a 100× objective lens. Scale bars are 5 μm. Platelet spreading on poly-l-lysine was quantified, and the percentage of totally spread platelets relative to the total adhered platelets is shown in the bar plot. Values are mean ± SD. Images were acquired in a Leica SP8 confocal microscope with a 63× objective lens (magnification, 2×). Scale bars are 20 μm. (D) Western blot pictures of β1-tubulin levels in platelet lysates from patients and controls, using 2 different β1-tubulin antibodies; β-actin was used as internal control.
As shown in Figure 7A-C, MKs forming proplatelets were rare, most were small and clustered, with shorter microtubules (Figure 7D). Although in both homozygous and heterozygous patients, MKs adhered and spread normally on fibrinogen, only in homozygous MKs was the β1-tubulin distribution aberrant, accumulating in small aggregates (Figure 7B). Also, the final platelet maturation in blood was defective, as indicated by the presence in blood of less barbell-proplatelets and “figure 8” structures and more preplatelets (supplemental Figure 6C).
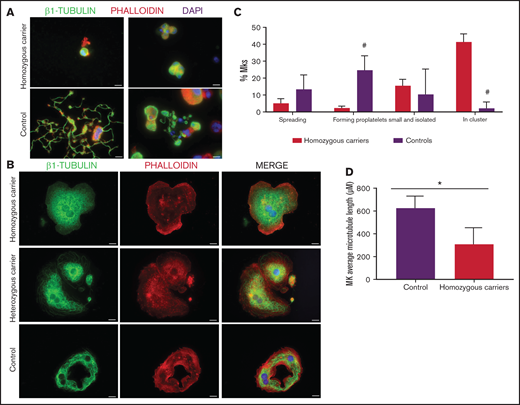
Effect of the TUBB1 p.Gly109Glu variant in CD34+ peripheral blood cell–derived MK cultures. (A) Representative images of CD34+ peripheral blood cell–derived MKs in 2 controls and in homozygous carriers (pedigree I) of the novel variant p.Gly109Glu. (B) Detailed images of spread MKs. MKs were labeled with β1-tubulin (green) and rhodamine-phalloidin (red). Nuclei were stained with DAPI (blue). Images were acquired in a Carl Zeiss Axio Observer.A1 fluorescence microscope with a 63× objective lens. Scale bars are 5μm. (C) Classification of MKs (as described in Figure 3; #P < .05) in homozygous carriers and controls. (D) Microtubule length in MKs from controls and homozygous carriers of the p.Gly269Asp β1-tubulin variant was assessed using ImageJ software. Values are mean ± SD of values obtained from at least 6 different microscopy fields. *P < .05.
Effect of the TUBB1 p.Gly109Glu variant in CD34+ peripheral blood cell–derived MK cultures. (A) Representative images of CD34+ peripheral blood cell–derived MKs in 2 controls and in homozygous carriers (pedigree I) of the novel variant p.Gly109Glu. (B) Detailed images of spread MKs. MKs were labeled with β1-tubulin (green) and rhodamine-phalloidin (red). Nuclei were stained with DAPI (blue). Images were acquired in a Carl Zeiss Axio Observer.A1 fluorescence microscope with a 63× objective lens. Scale bars are 5μm. (C) Classification of MKs (as described in Figure 3; #P < .05) in homozygous carriers and controls. (D) Microtubule length in MKs from controls and homozygous carriers of the p.Gly269Asp β1-tubulin variant was assessed using ImageJ software. Values are mean ± SD of values obtained from at least 6 different microscopy fields. *P < .05.
We also assessed α-tubulin distribution by immunofluorescence. Although α-tubulin and β1-tubulin in resting platelets from controls and heterozygous carriers localized in the tubulin ring (supplemental Figure 8A) with a strong colocalization (supplemental Figure 9), in platelets from homozygous β1-tubulin Glu109 variant carriers, the marginal band was mainly made of α-tubulin (supplemental Figures 7 and 8A). In spread platelets, α-tubulin incorporated into microtubules in controls and in all the β1 tubulin variant carriers (supplemental Figure 8B). Western blot analysis showed a minimal reduction in α-tubulin levels in homozygous carriers of this variant, whereas acetylated α-tubulin was increased in homozygous and, to a lesser extent, in heterozygous carriers (supplemental Figure 8C). Quantitative reverse transcription-polymerase chain reaction showed a marked reduction in TUBB1 mRNA levels in homozygous carriers, whereas the mRNA level of β5-tubulin (TUBB5), an isoform upregulated in β1-Tubb1−/− mice,14 was increased (supplemental Figure 8D).
Discussion
This study expands current knowledge on TUBB1-RT by reporting the largest series of families with this disorder and showing wide heterogeneity in genetic and clinical presentation. In 8 of the 9 studied pedigrees, we found heterozygous carriers of 5 variants in TUBB1, 4 of which (c.35del [p.Cys12LeufsTer12], c.319A>C [p.Thr107Pro], c.1267C>T [p.Gln423*], and c.1075C>T [p.Arg359Trp]) were previously reported by us24 but now are functionally characterized. The c.806G>A [p.Gly269Asp] TUBB1 variant is instead novel and reported here for the first time. In the ninth pedigree, we found the c.326G>A [p.Gly109Glu] variant. This variant was previously identified by genome-wide association studies in heterozygosis as a low-frequency gene variant (minor allele frequency = 0.087%) associated with low platelet count and increased MPV and platelet distribution width45 and more recently in 4 patients with a platelet defect enrolled in the ThromboGenomics project.26 However, here we report for the first time the presence of the p.Gly109Glu variant in homozygosity in a TUBB1-RT pedigree.
All variants but p.Gln423* were initially classified as VUS. The functional studies and segregation data described here allowed a reclassification of 4 variants as pathogenic or likely pathogenic (supplemental Table 3). Thus, our study underscores the importance of functional studies to correctly interpret the pathogenicity of TUBB1 variants. The appropriate curation of gene variants associated with diseases is considered very relevant and is currently underway by the Clinical Genome Resource (ClinGen) with the Variant Curation Expert Panels. Because, in contrast to Glanzmann thrombasthenia33 and RUNX1-related disease,34,35 interpretation rules for variants involved in TUBB1-RT have not been generated yet, it is highly warranted that this is made by a ClinGen expert panel.
Incomplete penetrance in TUBB1-RT has been overlooked thus far and was suggested only very recently in a large Chinese family carrying the TUBB1 p.Arg318Trp variant.27 In contrast, this phenomenon has been reported for common TUBB1 variants, which are considered only as modifiers of the thrombocytopenia phenotype caused by variants in other genes.15,18
Here, by segregation studies, we show incomplete penetrance also of rare TUBB1 variants for platelet traits: (1) 1 of 4 carriers of the p.Cys12LeufsTer12 (families A and B) presented with increased MPV but normal platelet count; (2) 1 p.Gln423* carrier (family E) displayed thrombocytopenia but normal platelet size; and (3) half of the carriers of p.Arg359Trp (families F and G) and one-third of the p.Gly269Asp carriers (family H) presented with normal platelets (Figure 1). In contrast to previous data,26,45 in our study only homozygous carriers of p.Gly109Glu displayed macrothrombocytopenia, whereas heterozygous carriers presented with only increased MPV or completely normal platelets. These data highlight the relevance of allele burden for the phenotypic expression of this disorder and support the notion that heritability of TUBB1-RT can be more complex than simply autosomal dominant.21,22,46
The fact that the higher levels of immature platelets were present in carriers of the missense variants with thrombocytopenia in respect to nonthrombocytopenic carriers (Table 1) may also support the hypothesis that additional factors contribute to modulating platelet turnover in pedigrees with TUBB1-RT. A potential limitation in determining IPF with automated hematology analysers is that mature platelets with large MPV, or a part of them, may be misclassified as immature platelets.
There seems to be a divergence in the impact of variants on β1-tubulin incorporation into microtubules and on platelet function. Transfection experiments in CHO cells and studies in patient platelets showed that Asp269, Pro107, and Glu109 β1-tubulin mutants, similar to p.Phe260Ser and p.Arg318Trp,22,23 did not incorporate into the microtubular network, whereas the p.Arg359Trp variant exerted a visible but weaker deleterious effect in β1-tubulin distribution. Abnormal tubulin distribution is a previously recognized finding in TUBB1-RT18,22,23,46 and supports pathogenicity of the variants found in our families.
Although further studies are required, in contrast to a recent high-throughput platelet spreading analysis,47 we found normal spreading of platelets and MKs from p.Arg359Trp heterozygous and p.Gly109Glu homozygous carriers, suggesting that β1-tubulin may not be critical for platelet spreading and contradicting the previous hypothesis of this protein being a key component of the spreading machinery.11,39
Immunoblotting showed that levels of β1-tubulin in platelets from p.Arg359Trp, p.Gly269Asp and p.Gly109Glu heterozygous carriers were minimally affected. In contrast, β1-tubulin was almost undetectable in platelet lysates from p.Gly109Glu homozygous carriers. At variance with our findings, reduced β1-tubulin in platelets has previously been reported in heterozygous carriers of the p.Arg318Trp and p.Phe260Ser TUBB1 variants,22,23 but the underlying mechanisms accounting for this were not elucidated. Although protein instability of mutant isoforms has been suggested,22,23 our data revealing lower TUBB1 mRNA levels in p.Gly109Glu homozygous carriers suggest that the β1 tubulin defect, at least in this family, may also be caused by defective TUBB1 transcriptional dysregulation.
A coordinated production of α- and β-tubulins is thought to be required for proper heterodimer assembly in cells.48,49 Indeed, heterozygous carriers of the p.Phe260Ser TUBB1 variant were shown to have markedly reduced platelet expression of both β1- and α-tubulin. In contrast, p.Gly109Glu homozygous carriers displayed almost unaffected platelet expression of α-tubulin, whereas acetylated α-tubulin, a marker of microtubule stability,50 and TUBB (β5-tubulin) mRNA levels were increased. These findings support the hypothesis that upregulation of other β-tubulin isoforms may counteract the β1-tubulin defect. Concordantly, we have previously shown in neonatal platelets that downregulation of β1-tubulin is counteracted by upregulation of other β-tubulin isoforms.39 Noteworthy, Tubb1−/− mice show overexpression of β2- and β5-tubulin, although these isoforms do not fully compensate for the absence of β1-tubulin.14 In addition, our finding of normal expression and localization of filamin, MYH9, DIAPH1, and α-actinin in platelets from carriers of the p.Gly109Glu variant indicates that, despite that components of the cytoskeleton are strictly connected and contribute to maintain cellular dynamics,51,52 their production is not influenced by β1-tubulin defect.
The deleterious functional effect of TUBB1 variants (p.Arg359Trp, p.Gly269Asp, and p.Gly109Glu) is strongly supported by the finding of impaired MK maturation and proplatelet formation by MKs differentiated from CD34+ cell from thrombocytopenic carriers of these mutations. Abnormal β1-tubulin localization in MKs and defective proplatelet formation have been previously observed for other TUBB1 variants.22,23,53,54 Additionally, we report for the first time that MKs from TUBB1-RT patients formed clusters in culture, as recently shown in DIAPH1-RD, an IT also associated to defects in cytoskeleton.55 Noteworthy, a very recent study of in vivo and in vitro proplatelet formation in mice has suggested that the β1-tubulin/microtubules axis plays an essential role in proplatelet formation in vitro, but is less critical for in vivo platelet release, which may in part explain the only moderate thrombocytopenia in patients with TUBB1-RT.56 Finally, we observed for the first time a defect of the late maturation steps of platelet generation from preplatelets. Twisting of microtubules to yield “figure 8” structures is crucial for the passage from preplatelets, large, oval-shaped circulating platelet precursors, into barbell-shapes and for subsequent separation of their extremes to obtain 2 mature platelets.40 It is therefore conceivable that a β1-tubulin defect may lead to reduced maturation of proplatelets contributing to thrombocytopenia.
Our study has some limitations: first, we cannot exclude that other variants in genes not present in our HTS panel or acquired conditions could contribute to thrombocytopenia in some patients; second, defective proplatelet formation is not a sufficient proof of the causality of TUBB1 variants in itself, and for this reason, we expanded these data with experiments performed with CHO cells transfected with the different variants.
In summary, our study expands the spectrum of TUBB1-RT (summarized in supplemental Table 4), showing a remarkable heterogeneity in clinical presentation, which suggests that allelic burden or combination with other genetic or phenotypic factors may modulate the effect of rare TUBB1 variants. In silico algorithms of prediction are not reliable enough when analyzing TUBB1 variants, ratifying the importance of clinical information and biological evaluation when establishing the pathogenicity of such variants. Our results may contribute to clarify the effect of β1-tubulin in platelet function and in thrombopoiesis.
Acknowledgments
The authors thank Antonio Moscardó (Centro de Investigación, Hospital La Fe, Valencia) for kindly providing anti-tubulin antibodies; Anabel Antón and Angel Esteban-Gil (IMIB-Arrixaca) for assistance with HTS performance and analysis; Francisca Ferrer-Marín (Centro Regional de Hemodonación) for helpful discussion; and Kristy Lee (UNC Chapel Hill, NC) for helpful advice in application of ACMG rules for variant classification.
The views expressed in this manuscript are those of the authors and do not necessarily represent the views of the National Heart, Lung, and Blood Institute; the National Institutes of Health; or the US Department of Health and Human Services.
This work was partially supported by grants from Instituto de Salud Carlos III (ISCIII) and Feder (PI17/01311, PI17/01966, PI20/00926 and CB15/00055), Fundación Séneca (19873/GERM/15), Gerencia Regional de Salud (GRS 2061A/19 and 1647/A/17), Fundación Mutua Madrileña (AP172142019), and Sociedad Española de Trombosis y Hemostasia (Premio López Borrasca 2019 and Ayuda a Grupos de Trabajo en Patología Hemorrágica). The authors’ research on inherited platelet disorders is conducted in accordance with the aims of the Functional and Molecular Characterization of Patients with Inherited Platelet Disorders Project, from Grupo Español de Alteraciones Plaquetarias Congénitas, which is supported by the Spanish Society of Thrombosis and Haemostasis. V.P.-B. has a predoctoral contract from CIBERER. L.B. was supported by a fellowship from Fondazione Umberto Veronesi. M.E.d.l.M.-B. holds a postdoctoral fellowship from the University of Murcia. A.M.-Q. holds a predoctoral grant from the Junta de Castilla y León.
Authorship
Contribution: V.P.-B., L.B., N.R., N.B., M.P.F.-P., A.M.-Q., M.E.d.l.M.-B., C.M., and J.R. performed platelet function studies; V.P.-B., L.B., and S.K. developed CHO models; V.P.-B., N.B., L.B., R.B., J.P., J.M.B., and J.R. performed molecular studies and HTS analysis; A.R.A., N.R., M.L.L., M.F.L.-F., S.M., V.V., P.G., J.M.B., and J.R. collected patient samples and interpreted clinical records; V.P.-B., L.B., S.K., C.M., P.G., M.L.L., J.M.B., and J.R. wrote the manuscript; and all authors helped in data interpretation and critically reviewed the paper.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: José Rivera, Centro Regional de Hemodonación-Universidad de Murcia, Ronda de Garay s/n, Murcia 30003, Spain; e-mail: jose.rivera@carm.es.

