

Key Points
Anti-BCMA CAR T cells caused a 7-month aplasia of bone marrow normal plasma cells and a longer period of hypogammaglobulinemia.
Prolonged hypogammaglobulinemia suggests a profound and lasting humoral immune deficiency after anti-BCMA CAR T-cell therapy.

Abstract
Systematic and dynamic humoral immune reconstitution is little-known for patients with relapsed/refractory (R/R) multiple myeloma (MM) who received anti–B-cell maturation antigen (BCMA) chimeric antigen receptor (CAR) T-cell therapy. We investigated the kinetics of B-cell, normal plasma cell, and immunoglobulin recovery in 40 patients who achieved ongoing response after anti-BCMA CAR T-cell therapy. All patients developed B-cell aplasia and the median duration of B-cell aplasia was 70 days (range, 23-270). The B-cell count reached its nadir on median day 7 and returned to baseline level on median day 97. BCMA+ cells in bone marrow turned undetectable on median day 28 (13-159) in 94.87% (37 of 39) of patients. Normal plasma cells in bone marrow were first redetected on median day 212. All patients developed a significant decrease in serum IgG, IgA, and IgM on median day 60. At year 1, recovery of serum IgG, IgM, and IgA was observed in 53.33% (8 of 15; non-IgG MM), 73.08% (19 of 26; non-IgM MM), and 23.81% (5 of 21;non-IgA MM) of the patients, respectively. Median time to IgG, IgM, and IgA recovery were days 386, 254, and not reached during follow-up, respectively. Virus-specific IgG levels decreased with loss of protection. Twenty-three of 40 (57.5%) patients had a total of 44 infection events. There were no infection-related deaths. These results reveal a 7-month aplasia of bone marrow normal plasma cells and longer period of hypogammaglobulinemia, suggesting a profound and lasting humoral immune deficiency after anti-BCMA CAR T-cell therapy, especially for IgA.
Introduction
Chimeric antigen receptor (CAR) T-cell therapy targeting B-cell maturation antigen (BCMA) has yielded encouraging results in treating relapsed/refractory (R/R) multiple myeloma (MM).1-4 Common acute toxicities, including cytokine release syndrome (CRS) and neurotoxicity, were taken seriously and were managed by treating with anti-interleukin-6 receptor blockade and/or corticosteroids in most situations.5,6 However, there are still some late adverse events, such as off-target effects, prolonged cytopenia, immune deficiency, and infections,2,7 which are becoming increasingly recognized.
BCMA is exclusively expressed on B-lineage cells, including plasmablasts and, in particular, at the stage from mature B-cell to plasma-cell terminal differentiation, as well as on malignant B cells and plasma cells, but is not expressed on naive and most memory B cells.8-11 Even though BCMA knockout mice showed normal B-cell development and no defects in short-term production of immunoglobulins and early humoral immune response,12 BCMA is critical for differentiation and survival of long-lived plasma cells in the bone marrow, which is essential for maintaining humoral immunity.10,13 The nearly ubiquitous BCMA-expression on myeloma cells makes it an ideal target for immunotherapy. All BCMA+ cells including normal plasma cells and myeloma cells are targeted by anti-BCMA CAR T cells and destroyed. Hence, the “on-target, off-tumor” activity of BCMA-specific CAR T cells eliminates normal plasma cells and causes hypogammaglobulinemia.14 Only sporadic reports4,7,14 showed immunosuppression in patients with R/R MM treated by anti-BCMA CAR T cells, and persistent hypogammaglobulinemia occurred in a few cases with a prolonged ongoing response. B-cell aplasia and hypogammaglobulinemia cause a high risk of infection, which may be a major cause of mortality in patients with MM.15 Therefore, the effect of anti-BCMA CAR T-cell therapy on humoral immune function deserves more investigation.
So far, little is known about systematic and dynamic humoral immune reconstitution in patients who have received anti-BCMA CAR T-cell therapy. We conducted a retrospective study to characterize the kinetics of B-cell, normal plasma cell, and immunoglobulin recovery and changes of antigen-specific antibodies in patients with R/R MM who achieved an ongoing response after anti-BCMA CAR T-cell therapy.
Methods
Patient selection
This study enrolled 40 patients with R/R MM who achieved a response after infusion of anti-BCMA CAR-T cells that incorporated either CD28- or 4-1BB-costimulated CARs.16,17 The patients were participants in clinical trials at the Affiliated Hospital of Xuzhou Medical University (Chinese trial registry chictr.org.cn #ChiCTR-OIC-17011272); Tongji Hospital of Tongji Medical College, Huazhong University of Science and Technology (chictr.org.cn #ChiCTR-OPC-16009113); and Tongji Hospital, Tongji University School of Medicine (clinicaltrials.gov #NCT04500431) from March 2017 through January 2020 (data cutoff, 15 November 2020). The study was approved by the ethics committees of the Affiliated Hospital of Xuzhou Medical University; Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology; and Tongji Hospital, Tongji University School of Medicine, in accordance with the principles of the Declaration of Helsinki.
CAR T-cell infusion and supportive treatment
Patients received lymphodepletion chemotherapy with fludarabine (30 mg/m2 per day; days −5 to −2) and cyclophosphamide (750 mg/m2; day −5) before T-cell infusion. Two different doses of anti-BCMA CAR T cells (1-2 × 106 and 1-2 × 107 CAR T cells per kilogram) were infused on day 0. Granulocyte colony-stimulating factor was administered when the neutrophil count was <0.5 × 109/L, until the count returned to normal. Intravenous IgG (IVIG) was used only in patients with IgG <400 mg/dL who also had frequent or severe infections. Antimicrobial prophylaxis was used when neutropenia developed after lymphodepletion and continued until the neutrophil count recovered or there was a need to change the therapy.
Clinical and laboratory data collection
Laboratory assessments, including complete blood counts, lymphocyte subsets, bone marrow plasma cells, and serum immunoglobulin concentrations were collected and compared with normal clinical laboratory values. B-cell and T-lymphocyte subsets, including CD4+ T cells, CD8+ T cells, and natural killer (NK) cells in peripheral blood, were detected by flow cytometry. Serum concentrations of IgG, IgA, and IgM were detected by immunonephelometry. Fluorescence-activated cell sorting was used to identify the persistence of CAR T cells, and the number of circulating CAR T cells per microliter was calculated on the basis of absolute CD3+ T-lymphocyte counts. The percentage of plasma cells present in the bone marrow was assessed in an immunophenotypic approach (flow cytometry). CD38 and CD27 were strongly expressed, and CD138 was expressed on the surface of normal plasma cells, which showed a normal partition of the κ and λ light chains. CD45 and CD19 were weakly expressed, whereas CD56 was not expressed on most normal plasma cells. Typical immunotyping of myeloma cells was characterized by abnormal expression of CD27, CD45, CD56, and CD19 and weak expression of CD38 and a display of immunoglobulin light-chain restriction, as previously reported.18
Clinical data, such as baseline characteristics of the patients, previous therapies, grades of CRS and neurotoxicity, infections, supportive treatments, and time to disease progression, death, other treatment, or last follow-up were also recorded. Evaluations of immune reconstitution and infections were performed once a month during the first 3 months after CAR T-cell infusion, then every 3 months until disease progression, subsequent therapy, or death. We censored the data at the time of disease progression or subsequent therapy from the analysis.
Antigen-specific antibody measurement
Antigen-specific antibody levels were measured by enzyme-linked immunosorbent assay kits, according to the manufacturer’s instructions, in patients who had available serum samples and had not received immunoglobulin replacement therapy. Only patients who had protective antibody levels before anti-BCMA CAR T-cell therapy were assessed. Serum measles-, rubella-, and mumps-specific IgG levels were retrospectively measured before lymphodepletion chemotherapy and at 1, 2, 3, 6, 9, and 12 months after anti-BCMA-CAR T-cell infusion. Thresholds of protective antibody were defined as >80 mIU/mL for measles, >240 mIU/mL for mumps, and >120 mIU/mL for rubella.
Evaluation criterion
The response of patients with R/R MM after CAR T-cell infusion was evaluated by the International Myeloma Working Group Revised Uniform Response (2014) criteria.19 The diagnosis and grading of CRS were made according to criteria proposed by Lee et al.5 B-cell aplasia was defined as <1% CD19+ peripheral blood mononuclear cells.20 Loss of CAR T-cell function was defined as BCMA expression that was redetectable in bone marrow by flow cytometry. Based on normal levels for IgG, IgM, and IgA in our laboratory, patients with serum IgG <800 mg/dL, IgM <50 mg/dL, and IgA <100 mg/dL were classified as having hypogammaglobulinemia.21 Infections included both definite (microbiologically diagnosed) and clinical (nonmicrobiologically diagnosed) infections. Infection severity was classified as mild, moderate, severe, life-threatening, or fatal, as previously established (supplemental Table 1).22
Statistical analysis
Quantitative data were expressed as the median (range). The Mann-Whitney U test and Kruskal-Wallis test were adopted for continuous variables. Fisher’s exact test and χ2 test were performed for categorical variables. The Spearman correlation test was used for correlation analysis. Cox proportional hazards modeling was used to examine the clinical and treatment factors in the recovery of B cells and immunoglobulins. Binary logistic regression models were performed to assess the factors associated with B-cell recovery at 3 months, undetectable IgA and IgM, and immunoglobulin recovery at 1 year. All variables achieving P < .15 in univariate analysis were considered for multivariate analysis. The Kaplan-Meier method was used to evaluate the association of variables with time to lymphocyte recovery, progression-free survival (PFS), and overall survival (OS). All statistical analyses were performed with IBM SPSS 25.0. Two-sided values reaching P < .05 were considered to be statistically significant.
Results
Clinical and treatment characteristics of patients
This study included 40 patients with R/R MM who received infusion of anti-BCMA CAR T cells, including 17 with the IgG subtype, 11 with the IgA subtype, and 12 with other subtypes (including 7 IgD and 5 light chain). The median follow-up time was 16 months (7-42). Patient clinical characteristics are shown in Table 1. Before anti-BCMA CAR T-cell infusion and lymphodepletion, 7.5% (3 of 40) of the patients had a normal B-cell count; 17.39% (4 of 23; non-IgG MM) had normal IgG concentrations, 10.34% (3 of 29; non-IgA MM) had normal IgA concentrations, and only 2.5% (1 of 40; non-IgM MM) had normal IgM concentrations. All 40 of the patients achieved a response, including 31 with complete response (CR)/stringent complete response (sCR); 5 with very good partial response; and 4 with partial response. CRS of any grade developed in 97.5% (39 of 40) of the patients; 15% (6 of 40) had grade 3 or 4. Kaplan-Meier curves of PFS and OS for all patients are shown in supplemental Figure 1.
Baseline and treatment characteristics
| Characteristic | All patients (n = 40) |
|---|---|
| Baseline characteristics | |
| Age, years, median (range) | 55 (27-70) |
| Male gender, n (%) | 27 (67.5) |
| Subtype (monoclonal globulin), n (%) | |
| IgG | 17 (42.5) |
| IgA | 11 (27.5) |
| IgD | 7 (17.5) |
| Light chain | 5 (12.5) |
| ISS stage, n (%) | |
| I | 15 (37.5) |
| II | 13 (32.5) |
| III | 12 (30) |
| Previous therapy lines, median (range) | 4 (2-9) |
| CAR T-cell costimulatory molecule, n (%) | |
| CD28 | 11 (27.5) |
| 4-1BB | 29 (72.5) |
| Treatment characteristics | |
| CAR T-cell dose, n (%) | |
| (1-2) ×106 cells per kilogram | 21 (52.5) |
| (1-2) ×107 cells per kilogram | 19 (47.5) |
| CRS grade, n (%) | |
| No grade | 1 (2.5) |
| 1-2 | 33 (82.5) |
| 3-5 | 6 (15) |
| Neurotoxicity grade, n (%) | |
| No grade | 28 (70) |
| 1-2 | 12 (30) |
| 3-5 | 0 |
| Corticosteroids, n (%) | 12 (30) |
| Tocilizumab, n (%) | 11 (27.5) |
| Response, n (%) | |
| CR/sCR | 31 (77.5) |
| VGPR | 5 (12.5) |
| PR | 4 (10) |
| Characteristic | All patients (n = 40) |
|---|---|
| Baseline characteristics | |
| Age, years, median (range) | 55 (27-70) |
| Male gender, n (%) | 27 (67.5) |
| Subtype (monoclonal globulin), n (%) | |
| IgG | 17 (42.5) |
| IgA | 11 (27.5) |
| IgD | 7 (17.5) |
| Light chain | 5 (12.5) |
| ISS stage, n (%) | |
| I | 15 (37.5) |
| II | 13 (32.5) |
| III | 12 (30) |
| Previous therapy lines, median (range) | 4 (2-9) |
| CAR T-cell costimulatory molecule, n (%) | |
| CD28 | 11 (27.5) |
| 4-1BB | 29 (72.5) |
| Treatment characteristics | |
| CAR T-cell dose, n (%) | |
| (1-2) ×106 cells per kilogram | 21 (52.5) |
| (1-2) ×107 cells per kilogram | 19 (47.5) |
| CRS grade, n (%) | |
| No grade | 1 (2.5) |
| 1-2 | 33 (82.5) |
| 3-5 | 6 (15) |
| Neurotoxicity grade, n (%) | |
| No grade | 28 (70) |
| 1-2 | 12 (30) |
| 3-5 | 0 |
| Corticosteroids, n (%) | 12 (30) |
| Tocilizumab, n (%) | 11 (27.5) |
| Response, n (%) | |
| CR/sCR | 31 (77.5) |
| VGPR | 5 (12.5) |
| PR | 4 (10) |
ISS, International Staging System; PR, partial response; VGPR, very good partial response.
Kinetics of B-cell count in peripheral blood and plasma cells in bone marrow
All patients developed B-cell aplasia, and the median duration of the aplasia was 70 days (23-270) after CAR T-cell infusion. B-cell aplasia at 1-, 2-, and 3- months was observed in 95%, 57.5%, and 30% of patients, respectively. The B-cell count reached its nadir on median day 7 (0-21) and were significantly lower than those at baseline (P < .001). The B-cell count remained depressed up to 2 months after CAR T-cell infusion (Figure 1A). Median time of B-cell count returned to baseline level was on day 79 and returned to normal level on day 177 (Figure 1B-C). Individual subject curves of B-cell count are shown in supplemental Figure 2A.
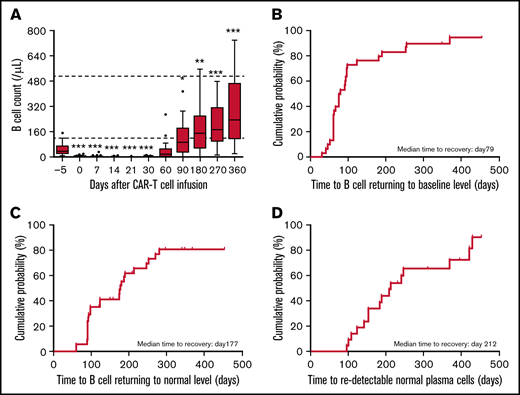
Kinetics of B cells and normal plasma cell recovery in patients treated with anti-BCMA CAR T cells. (A) Median counts of B lymphocytes at different time points before and after CAR T-cell infusion. Box-and-whisker plots show the median (horizontal line) and interquartile range (box). Dotted lines indicate the normal range. (B-C) Cumulative incidence of B-cell count returning to baseline (B) and normal (C) levels after anti-BCMA CAR T-cell therapy. (D) Cumulative incidence of normal plasma cells redetected in bone marrow after anti-BCMA CAR T-cell therapy. The significance of results was calculated by the Kruskal-Wallis test. *P < .05, **P < .01, ***P < .001, compared with baseline pretherapy levels.
Kinetics of B cells and normal plasma cell recovery in patients treated with anti-BCMA CAR T cells. (A) Median counts of B lymphocytes at different time points before and after CAR T-cell infusion. Box-and-whisker plots show the median (horizontal line) and interquartile range (box). Dotted lines indicate the normal range. (B-C) Cumulative incidence of B-cell count returning to baseline (B) and normal (C) levels after anti-BCMA CAR T-cell therapy. (D) Cumulative incidence of normal plasma cells redetected in bone marrow after anti-BCMA CAR T-cell therapy. The significance of results was calculated by the Kruskal-Wallis test. *P < .05, **P < .01, ***P < .001, compared with baseline pretherapy levels.
Patients receiving tocilizumab appeared to be more likely to achieve B-cell recovery in univariate analysis (adjusted hazards ratio [HR], 2.32; P = .043), although age, sex, International Staging System stage, previous therapy lines, costimulatory molecule of CARs, CAR T-cell dose, CRS grade, and administration of corticosteroids did not correlate with B-cell recovery (supplemental Table 2). The association between administration of tocilizumab and B-cell recovery (HR, 2.53; P = .037) remained significant in multivariate analyses. Furthermore, patients receiving fewer lines of therapy appeared to be more likely to have a normal B-cell count at 3 months in both univariate and multivariate analyses (P = .043 and .040, respectively; supplemental Table 3). Individual B-cell counts correlated with absolute lymphocyte count (ALC) and with CD4+ T-, CD8+ T-, and NK-cell counts at all time points (supplemental Figure 3A-D), but especially with ALC (Spearman correlation, r = 0.439; P < .0001; supplemental Figure 3A).
BCMA+ cells including normal plasma cells and myeloma cells in bone marrow turned undetectable on median day 28 (13-159) in 94.87% (37 of 39) of patients after anti-BCMA CAR T-cell infusion. Of those, 31 patients were available for continuous monitoring of BCMA expression. The median duration of persistent undetectable BCMA was 218 days (70-418), which was consistent with a prolonged persistence of anti-BCMA CAR T-cells, with 54.84% of the patients having detectable CAR T cells at 6 months as shown in supplemental Figure 4. Myeloma cells were redetectable in 10 patients at the time of relapse with BCMA positivity cells and no detectable normal plasma cells after CAR T-cell infusion. Normal plasma cells in bone marrow were first redetected on median day 212 in the remaining 21 patients (including 5 patients maintaining undetected BCMA throughout the follow-up; Figure 1D). Longer persistence of CAR T cells was associated with slower recovery of plasma cells, but not of B-cell count (supplemental Table 4).
Kinetics of serum immunoglobulin concentrations
Eight (20%) patients received IVIG supportive treatment after CAR T-cell infusion. The maximum dose was 20 g for each treatment and the shortest interval between administrations was 2 months. The serum for measuring IgG, IgM, and IgA concentrations was collected before IVIG. In view of the half-life of IgG, the dose, and the administration interval, we concluded that the influence of IVIG on detectable immunoglobulins could be ignored.
Serum IgG (in patients with non-IgG MM), IgM (in patients with non-IgM MM), and IgA (in patients with non-IgA MM) decreased significantly after CAR T-cell infusion compared with baseline prechemotherapy concentrations and reached nadir at day 60 (Figure 2A,C,E). Recovery of serum IgG to a normal level was observed in 53.33% (8 of 15) of patients (non-IgG MM) at 1 year. The median time to IgG recovery was day 386 (Figure 2A-B). Compared with IgG, IgM concentrations recovered more rapidly: 73.08% (19 of 26) of patients (non-IgM MM) recovered to a normal level at 1 year. The median time to IgM recovery was day 254 (Figure 2C-D). Serum IgM was undetectable in 52.5% (21 of 40) of patients (in 7, before lymphodepleting chemotherapy) on median day 30 (0-90) after CAR T-cell infusion. Factors related to undetectable IgM were evaluated, and a significant association with 4-1BB integrated CAR T cells was observed (P = .038; supplemental Table 5). Serum IgA concentration was the slowest to recover, with normal levels detected in only 23.81% (5 of 21) of patients (non-IgA MM) at 1 year. The median time to IgA recovery to normal levels was not reached during follow-up (Figure 2F). Serum IgA was undetectable in 79.3% (23 of 29) patients (in 5, before lymphodepleting chemotherapy) on median day 30 (7-180) after CAR T-cell infusion. Factors related to undetectable IgA were also evaluated, and no significant factors were identified (supplemental Table 6). Curves of serum IgG, IgM, and IgA concentrations for individual patients are shown in supplemental Figure 2B-D.
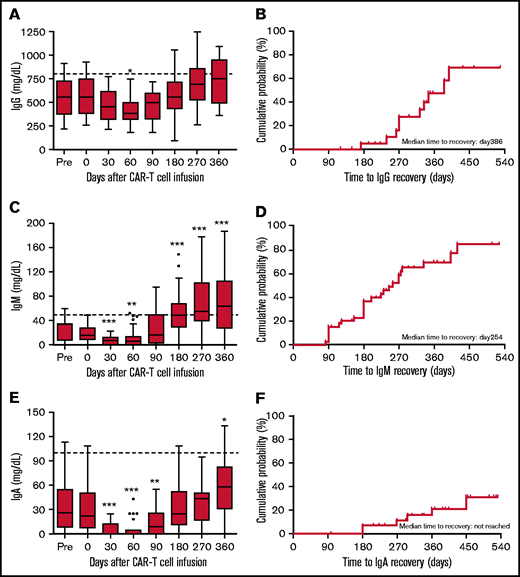
Kinetics of serum IgG, IgM, and IgA recovery in patients treated with anti-BCMA CAR T cells. (A) Box-and-whisker plots of IgG concentrations in patents with non-IgG MM at different time points before and after CAR T-cell infusion. (B) Cumulative incidence of IgG recovery. (C) Box-and-whisker plots of IgM concentrations in patients with non-IgM MM at different time points before and after CAR T-cell infusion. (D) Cumulative incidence of IgM recovery. (E) Box-and-whisker plots of IgA concentrations in patients with non-IgA MM at different time points before and after CAR T-cell infusion. (F) Cumulative incidence of IgA recovery. In panels A, C, and E, the median (bar) and interquartile range (box) are shown. The dotted line indicates the lower limit of normal. The significance of results are calculated by the Kruskal-Wallis test. *P < .05, **P < .01, ***P < .001, compared with baseline pretherapy levels.
Kinetics of serum IgG, IgM, and IgA recovery in patients treated with anti-BCMA CAR T cells. (A) Box-and-whisker plots of IgG concentrations in patents with non-IgG MM at different time points before and after CAR T-cell infusion. (B) Cumulative incidence of IgG recovery. (C) Box-and-whisker plots of IgM concentrations in patients with non-IgM MM at different time points before and after CAR T-cell infusion. (D) Cumulative incidence of IgM recovery. (E) Box-and-whisker plots of IgA concentrations in patients with non-IgA MM at different time points before and after CAR T-cell infusion. (F) Cumulative incidence of IgA recovery. In panels A, C, and E, the median (bar) and interquartile range (box) are shown. The dotted line indicates the lower limit of normal. The significance of results are calculated by the Kruskal-Wallis test. *P < .05, **P < .01, ***P < .001, compared with baseline pretherapy levels.
In multivariate analyses, clinical and treatment factors were found not to be associated with the recovery of serum IgG, IgM, and IgA concentrations (supplemental Table 7). However, patients in whom serum IgA concentrations returned to normal levels at 1 year had received fewer therapy lines and had a shorter duration of undetectable BCMA in bone marrow than patients whose IgA did not return to normal levels (P = .005 and .029, respectively; supplemental Table 8). Multivariate analysis was not performed as most P values were more than .15 in univariate analysis. Furthermore, a longer persistence of CAR T cells was associated with a slower recovery of IgA concentrations, but not of IgG or IgM (supplemental Table 4). Concentrations of serum IgG, IgM, and IgA all correlated with B-cell count (Spearman correlation, r = 0.288, 0.449, and 0.363; respectively, P < .0001; supplemental Figure 3E-G).
Dynamics of antigen-specific antibody levels
We analyzed antigen-specific IgG concentrations in 21 patients; only 3 (14.29%) had protective antimeasles or antimumps IgG levels and 4 (19.05%) had protective antirubella IgG levels before CAR T-cell infusion. Kinetics of antigen-specific IgG levels of these patients are presented in Figure 3. Measles-, rubella-, and mumps-specific IgG levels decreased with loss of protection in all the 3 or 4 patients after anti-BCMA CAR T-cell infusion. Antimeasles IgG levels in 1 of the 3 patients returned above the protective thresholds during the follow-up. Antirubella IgG levels showed a trend of recovery, and returned to protective thresholds in 1 of the 4 patients. Antimumps IgG levels remained below the minimum protective titer for a long time during the follow-up.
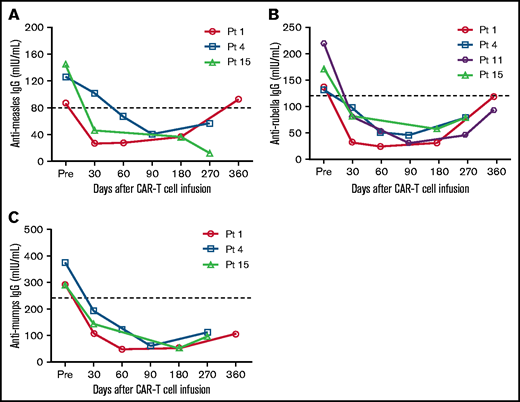
Antigen-specific IgG levels before and after anti-BCMA CAR T-cell infusion. Antirubella (A), antimumps (B), and antimeasles (C) IgG levels were measured in patients who had protective antibody levels before anti-BCMA CAR T-cell therapy. Thresholds of protective IgG levels are indicated by the dashed lines.
Antigen-specific IgG levels before and after anti-BCMA CAR T-cell infusion. Antirubella (A), antimumps (B), and antimeasles (C) IgG levels were measured in patients who had protective antibody levels before anti-BCMA CAR T-cell therapy. Thresholds of protective IgG levels are indicated by the dashed lines.
Humoral immunity reconstitution after loss of anti-BCMA CAR T-cell function
In the 31 patients available for continuous monitoring of BCMA expression, 6 presented loss of CAR T-cell function and were disease free and did not receive further therapy for more than 6 months after CAR T-cell loss, permitting the evaluation of long-term immune reconstitution. Five of the 6 patients experienced B-cell recovery before loss of anti-BCMA CAR T-cell function, and 1 patient still had B-cell depletion at the time of last follow-up. In the 6 patients, the M-component type was IgG in 1 case, IgA in 3 cases, IgD in 1 case, and light chain in 1 case. Three of 5 patients with non-IgG MM showed unrecovered IgG concentrations and 2 had recovered IgG at 6 and 3.6 months after loss of CAR T-cell function. One of 3 patients with non-IgA MM showed recovered IgA concentrations at 11.7 months after loss of CAR T-cell function. Interestingly, 4 of 6 patients with non-IgM MM had IgM recovery at the time of detectable BCMA (supplemental Table 9).
Infection events
Of the 40 patients, 30 (75%) received granulocyte colony-stimulating factor to treat neutropenia after CAR T-cell infusion; 27 (67.5%) patients received antibacterial or antifungal prophylaxis during the period of neutropenia. Twenty-three of 40 (57.5%) patients had a total of 44 infection events: 47.7% (21 of 44) were severe infections and 2.3% (1 of 44) were life-threatening infections, but there were no infection-related deaths. The most common microorganism was bacterial (n = 25), followed by viral (n = 8) and fungal (n = 3; Figure 4A). The most common infection sites were lung (n = 23), upper respiratory tract (n = 9), intestinal tract (n = 4), and bloodstream (n = 4; Figure 4B); 43.18% (19 of 44) of infections occurred between days 0 and 60 after CAR T-cell infusion. The incidence of infections between days 0 and 60 was higher than that between days 60 and 120, 120 and 180, 180 and 270, and 270 and 360 and at day 360 or later (15.91% [7 of 44], 15.91% [7 of 44], 11.36% [5 of 44], 9.09% [4 of 44], and 4.55% [2 of 44], respectively; Figure 4C). Serum IgG, IgM, and IgA concentrations during infection were analyzed. Patients with severe/life-threatening infections had lower serum IgG concentrations than patients with mild/moderate infections (Figure 4D), whereas serum IgM or IgA concentrations were comparable between patients with severe/life-threatening and mild/moderate infections (Figure 4E-F). Patient 13, who had sustained low concentrations of IgA and IgM, experienced persistent and repeated diarrhea from 6 to 14 months after CAR T-cell infusion. The characteristics of the infections, including severity, microorganism, and site, are shown for different periods in supplemental Table 10.
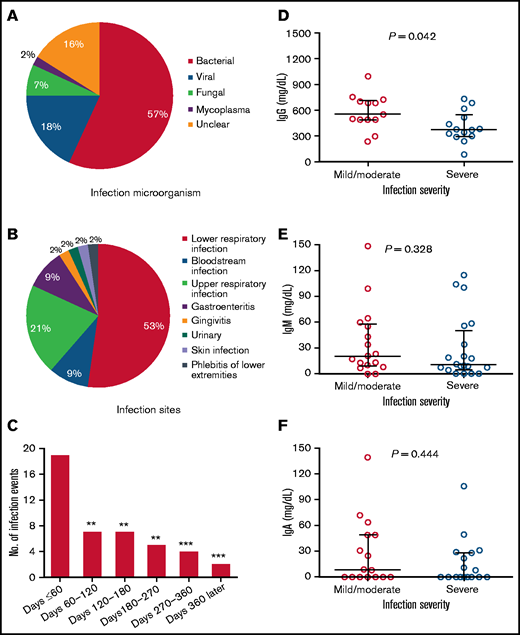
Infections after anti-BCMA CAR T-cell infusion. (A) Type of infectious microorganisms after anti-BCMA CAR T-cell infusion. (B) Infection sites after anti-BCMA CAR T-cell infusion. (C) Number of infections during different periods of time after CAR T-cell infusion. **P < .01, ***P < .001, compared with number of infections that occurred within 60 days after CAR T-cell therapy. (D) Serum IgG concentration during infection in patients with non-IgG MM with mild/moderate or severe/life-threatening infections. (E) Serum IgM concentration during infection in patients with non-IgM MM with mild/moderate or severe/life-threatening infections. (F) Serum IgA concentration during infection in patients with non-IgA MM with mild/moderate of severe/life-threatening infections. (D-F) Each point represents a single value from 1 person. Horizontal lines indicate median and interquartile range. The significance of the results were calculated by the Kruskal-Wallis test.
Infections after anti-BCMA CAR T-cell infusion. (A) Type of infectious microorganisms after anti-BCMA CAR T-cell infusion. (B) Infection sites after anti-BCMA CAR T-cell infusion. (C) Number of infections during different periods of time after CAR T-cell infusion. **P < .01, ***P < .001, compared with number of infections that occurred within 60 days after CAR T-cell therapy. (D) Serum IgG concentration during infection in patients with non-IgG MM with mild/moderate or severe/life-threatening infections. (E) Serum IgM concentration during infection in patients with non-IgM MM with mild/moderate or severe/life-threatening infections. (F) Serum IgA concentration during infection in patients with non-IgA MM with mild/moderate of severe/life-threatening infections. (D-F) Each point represents a single value from 1 person. Horizontal lines indicate median and interquartile range. The significance of the results were calculated by the Kruskal-Wallis test.
Correlation of B-cell and immunoglobulin recovery with PFS after CAR T-cell infusion
Patients were segregated into groups with PFS <1 year and ≥1 year. Immune reconstitution including B-cell, IgG, IgM, and IgA recovery were compared between the 2 groups. Three patients with ongoing CR/sCR were excluded because their follow-up times were less than 1 year. Patients (non-IgM MM) whose PFS were ≥1 year had a higher baseline B-cell count on day −5 (P = .007) and IgM concentrations on day 180 (P = .021) than patients whose PFS were <1 year (supplemental Figure 5). We also analyzed PFS based on the patients’ serum IgG, IgM, and IgA concentrations at 1 year. Patients (non-IgG MM) with recovered IgG concentrations at 1 year had a similar PFS compared with patients without (supplemental Figure 6A). Patients with recovered IgM (non-IgM MM) concentrations at 1 year after CAR T-cell infusion seemed to have a longer PFS (median, 1094 days) than patients with unrecovered IgM concentrations (median, 404 days). However, the difference was not statistically significant. A similar result was seen in IgA concentrations in patients with non-IgA MM (supplemental Figure 6B-C). Moreover, patients with a duration of CAR T-cell persistence of ≥6 months seemed to have a longer PFS (median, 1096 days) than patients with a duration of <6 months (median, 360 days). However, the difference was not significant (P = .256).
Discussion
Anti-BCMA CAR T-cell therapy has made a breakthrough in treating R/R MM, despite the accompanying acute toxicities. However, potential long-term adverse events, such as prolonged cytopenia and immune deficiency, as well as infections, have raised widespread concern.23-25 To our knowledge, this is the first systematic study to analyze the kinetics of B-cell and plasma cell aplasia and hypogammaglobulinemia after anti-BCMA CAR T-cell therapy in patients with R/R MM, and the results may facilitate the management of infectious complications in patients with long-term follow-up.
BCMA is exclusively expressed on plasma cells and is critical for differentiation and survival of long-lived plasma cells but is dispensable for B-cell homeostasis.10,13 Expression of BCMA was absent in naive B cells and peripheral blood memory B cells,26 CAR T-cell targeting of BCMA is thought to mainly lead to a decrease in plasma cells in bone marrow. In our study, the B-cell count decreased soon after lymphodepleting chemotherapy and recovered to baseline level on day 79 after anti-BCMA CAR T-cell infusion. Undetectable BCMA in bone marrow persisted for ∼7 months after CAR T-cell infusion, which was longer than the duration of B-cell aplasia. Fludarabine+cyclophosphamide induced a rapid and sustained reduction of all lymphocyte subsets.27,28 Our results showed that the B-cell count positively correlated with ALC, CD4+T-, CD8+ T-, and NK-cell counts, especially ALC, suggesting that B-cell depletion is probably not only caused by anti-BCMA CAR T cells but also by lymphodepleting chemotherapy.
Plasma cells are most recognizable for their ability to secrete large amounts of antibodies, thus positioning this cell type as a key component of humoral immunity.29 In our clinical trial, after anti-BCMA CAR T-cell infusion, all patients showed decreased serum IgG, IgM, and IgA concentrations. Serum IgM and IgA concentrations were undetectable in more than half of the patients. IgM concentration was the first to recover, followed by IgG and finally IgA. IgM was the earliest immunoglobulin synthesized and secreted into the serum, which is consistent with IgM showing the fastest recovery among the 3 types of immunoglobulins after CAR T-cell therapy. More patients who received 4-1BB–integrated CAR T cells had undetectable IgM levels than patients who received CD28-integrated CAR T cells in our study, which may be related to the longer persistence of CAR product with 4-1BB costimulation. However, studies with larger sample size should be conducted to investigate these issues. Upon binding to its cognate ligand, a proliferation-inducing ligand, BCMA, induces immunoglobulin isotype switching, especially IgA production.30 Perhaps anti-BCMA CAR T cells caused the slowest recovery of IgA by inhibiting isotype switching of IgA, which may explain the association between longer persistence of CAR T-cell and slower recovery of IgA. Patients with recovered IgA concentrations at 1 year had shorter duration of undetectable BCMA expression in bone marrow, which may be explained by the role of BCMA on immunoglobulin isotype switching. In our study, 6 patients presented with loss of CAR T-cell function and were disease free and did not receive further therapy for >6 months after CAR T-cell loss. Despite B-cell recovery and loss of CAR T-cell function, patients still showed an impairment of immunoglobulin production of differing severity, suggesting profound and lasting hypogammaglobulinemia after anti-BCMA CAR T-cell therapy.
BCMA is selectively expressed by long-lived plasma cells,11 which secrete antibodies that participate in permanent immunity. Measles-, rubella-, and mumps-specific IgG were measured to reflect humoral immunity caused by the high incidence of seropositivity and the stability of virus-specific IgG over time in healthy individuals.31 In our study, measles-, rubella-, and mumps-specific IgG levels decreased with loss of protection, which indicated that preexisting humoral immunity was impaired by anti-BCMA CAR T-cell therapy. Antigen-specific IgG levels remained below the minimum protective titer for a long time during the follow-up, reflecting lasting humoral immune deficiency that may increase the risk of infections in anti-BCMA CAR T cell–treated patients with R/R MM.
Infections are attributable to factors including immune dysfunction caused by disease per se, effects of previous antitumor therapies, lymphodepleting chemotherapy, corticosteroids and tocilizumab for treating CRS, and prolonged cytopenia and hypogammaglobulinemia related to CAR T-cell–mediated cytotoxicity.32-34 Prolonged persistence of anti-BCMA CAR T cells is beneficial for immunosurveillance against myeloma cells, but it also eliminates normal plasma cells, which increases the risk of humoral immune deficiency and infections. The incidence of infections including severe infections in anti-BCMA CAR T-cell–treated patients with R/R MM seems higher than that in anti-CD19 CAR T-cell–treated patients with lymphoma,24 which may be relevant to both myeloma itself and CAR T-cell’s targeting effects. Secretory IgA and IgM play an important role in mucosal immunity and preventing local infection at the mucosal surface, especially in the upper respiratory and gastrointestinal tracts,35,36 which may explain the persistent and repeated diarrhea that patient 13, who had sustained low serum IgA and IgM concentrations, experienced during the long follow-up.
Patients with severe/life-threatening infections had lower IgG concentrations than patients with mild/moderate infections, confirming the presence of lasting and profound hypogammaglobulinemia and related severe infections after anti-BCMA CAR T-cell infusion. Considering the long-term deficiency of plasma cell and hypogammaglobulinemia after anti-BCMA CAR T-cell therapy, IVIG is necessary.37 In our study, IVIG was used only in patients with IgG <400 mg/dL who also had frequent or severe infection. Although the incidence of severe infections was not low, no infection-related deaths occurred, and life-threatening infections were rare. Therefore, we suggest that patients with severe hypogammaglobulinemia, who also have frequent or severe infections, receive immunoglobulin replacement therapy after anti-BCMA CAR T-cell therapy; such supporting treatments may have to be continued for several months or longer. Because of the small sample size in our study, we cannot assess the correlation between immunoglobulins and specific types of infections.
Many myeloma therapies are toxic to lymphocytes and affect functional capacity of both T and B cells, which may have an influence on the efficacy and durability of CAR T-cell therapy. Therefore, patients with a longer PFS had higher baseline B-cell counts, probably because they had fewer lines of previous therapy and less intensive recent myeloma therapy. Gonzalez-Calle et al38 confirmed that long-term recovery of polyclonal immunoglobulin production has been associated with improved survival outcomes. In our trial, patients whose PFS was ≥1 year had higher IgM concentrations at day 180 than those who relapsed within 1 year, which may suggest a prognostic role of immunoglobulin recovery in PFS. However, the recovery of immunoglobulins suggests loss of CAR T-cell activity and would be expected to result in relapse. Further results showed no significant difference of PFS between patients with recovered IgM concentrations at 1 year and patients without IgM recovery. Whether recovery of IgM may serve as a supplementary prognostic marker for patients with R/R MM after anti-BCMA CAR T-cell therapy deserves further study.
In summary, B-cell aplasia occurred soon after lymphodepleting chemotherapy and remained depressed up to 2 months after anti-BCMA CAR T-cell therapy. Anti-BCMA CAR T-cells caused a 7-month aplasia of normal bone marrow plasma cells and a longer period of hypogammaglobulinemia. Prolonged hypogammaglobulinemia suggests a profound and lasting humoral deficiency, especially of IgA, after anti-BCMA CAR T-cell therapy. IVIG should be given in patients with severe hypogammaglobulinemia who also have frequent or severe infections and may have to be continued for several months or longer.
Acknowledgments
The authors thank all the faculty members and staff in the Clinical and Laboratory Unit of the Department of Hematology, the Affiliated Hospital of Xuzhou Medical University; Tongji Hospital of Tongji Medical College, Huazhong University of Science and Technology, Tongji Hospital, Tongji University School of Medicine, and Xinqiao Hospital, Army Medical University for their generous assistance and support.
This work was supported in part by the Program of National Natural Science Foundation of China (grants 81930005, 81970159, and 81700177) and the Research and Practice Innovation Program for Postgraduates of Jiangsu Province (KYCX20_2457).
Authorship
Contribution: K.X., J. Zhou, and A.L. designed the research; H.C., F.Z., H.S., W.S., D.L., and Z.Y. and their respective research teams recruited and followed up the patient and provided clinical data of patients; Y.W., C.L., J.X., and J.C. collected and analyzed research data; P.L., H.L., K.Q., X.W., and X.T. performed statistical analyses; G.J. contributed the CAR T cells of this study; Y.W., B.P., and K.X. drafted and revised the manuscript; X.Z., J. Zheng, and Z.L. provided support in the project; all authors contributed to the writing and revision of the manuscript and approved the final version.
Conflicts-of-interest disclosure: G.J. is an employee of iCARTAB Biomedical Co Ltd. The remaining authors declare no competing interests.
Correspondence: Kailin Xu, Department of Hematology, The Affiliated Hospital of Xuzhou Medical University, 99 West Huaihai Rd, Xuzhou 221002, China; e-mail: lihmd@163.com; Jianfeng Zhou, Department of Hematology, Tongji Hospital of Tongji Medical College, Huazhong University of Science and Technology, Wuhan, Hubei 430030, China; e-mail: jfzhou@tjh.tjmu.edu.cn; and Aibin Liang, Department of Hematology, Tongji Hospital, Tongji University School of Medicine, Shanghai 200065, China; e-mail: lab7182@tongji.edu.cn.

