

Key Points
FBXO11 loss-of-function mutations are frequent in BL, contributing to increased BCL6 stabilization and lymphoma progression.
Specific targeting of BCL6 is a valuable therapeutic option for BL.

Abstract
The expression of BCL6 in B-cell lymphoma can be deregulated by chromosomal translocations, somatic mutations in the promoter regulatory regions, or reduced proteasome-mediated degradation. FBXO11 was recently identified as a ubiquitin ligase that is involved in the degradation of BCL6, and it is frequently inactivated in lymphoma or other tumors. Here, we show that FBXO11 mutations are found in 23% of patients with Burkitt lymphoma (BL). FBXO11 mutations impaired BCL6 degradation, and the deletion of FBXO11 protein completely stabilized BCL6 levels in human BL cell lines. Conditional deletion of 1 or 2 copies of the FBXO11 gene in mice cooperated with oncogenic MYC and accelerated B-cell lymphoma onset, providing experimental evidence that FBXO11 is a haploinsufficient oncosuppressor in B-cell lymphoma. In wild-type and FBXO11-deficient BL mouse and human cell lines, targeting BCL6 via specific degraders or inhibitors partially impaired lymphoma growth in vitro and in vivo. Inhibition of MYC by the Omomyc mini-protein blocked cell proliferation and increased apoptosis, effects further increased by combined BCL6 targeting. Thus, by validating the functional role of FBXO11 mutations in BL, we further highlight the key role of BCL6 in BL biology and provide evidence that innovative therapeutic approaches, such as BCL6 degraders and direct MYC inhibition, could be exploited as a targeted therapy for BL.
Introduction
BCL6 is a transcriptional repressor that is frequently deregulated in human lymphoma.1 By binding to specific DNA sequences, BCL6 controls the transcription of a variety of genes involved in B-cell development, differentiation, and activation.1 Several genetic mechanisms contribute to BCL6 deregulation, including chromosomal translocations,2 mutations in its promoter region that affect BCL6 transcription,1 and defects in acetylation-mediated inactivation of BCL6.3 Increased expression of BCL6 results in a block of lymphoid cell differentiation, as well as resistance to genomic damage and apoptosis.4,5 The ubiquitin ligase FBXO11 targets BCL6 for ubiquitylation and subsequent degradation. Mutations or deletions of FBXO11 represent an additional mechanism for deregulation of BCL6 expression in diffuse large B-cell lymphoma (DLBCL)6 ; however, they are poorly characterized in other lymphoma types.
FBXO11 expression is not restricted to lymphoid tissue; in fact, it is ubiquitous. Additional targets of FBXO11 ubiquitin ligase activity have recently been described that include CDT2,7,8 p53,9-11 SNAIL,12 and BLIMP-1.13,14 A neurodevelopmental disorder with variable intellectual disability and other phenotypic features (MIM 618089) has recently been associated with loss-of-function mutations of FBXO11.15-17 The mouse mutant Jeff carries a missense mutation in the FBXO11 gene.18 FBXO11 is a 930-aa-long F-box protein that contains a proline-rich domain and the F-box domain at the N terminus, 3 central carbohydrate-binding proteins and sugar hydrolyses (CASH) domains, and a zinc finger domain at the C terminus. The functional domains of the protein are the CASH regions that are responsible for substrate recognition, whereas the F-box domain is essential for binding the SKP1 protein of the ligase complex.6,19
Knockout (KO) of FBXO11, specifically in germinal centers (GCs) in mice, results in higher levels of BCL6 protein in GCs and an increased number of GC B cells, eventually leading to the development of lymphoproliferative disorders, but not overt lymphoma, after a long latency of 15 to 18 months.20 In GC cells, FBXO11 supports CD40 expression by targeting repressors BCL6 and CTBP1; FBXO11 KO decreases primary B-cell CD40 abundance and impairs class-switch recombination.21 Thus, although FBXO11 has been proposed to act as a haploinsufficient tumor-suppressor gene, experimental validation of its role as an oncosuppressor is lacking.
Burkitt lymphoma (BL) typically expresses BCL6, and recently reported studies showed frequent FBXO11 mutations.22-24 Therefore, we molecularly and biologically characterized the role of FBXO11 in BL. All mutations showed an impaired ability to regulate BCL6 and SNAIL degradation, thus indicating that FBXO11 mutations in BL are loss-of-function mutations. Deletion of 1 or both FBXO11 alleles strongly accelerated the development of lymphoma in Eμ-Myc mice. In human BL lines, FBXO11 deletion almost completely stabilized BCL6 expression, and targeting BCL6 in BL lines using BCL6 inhibitors or BCL6 degraders impaired BL growth in vitro and in vivo, thus identifying BCL6 as an actionable therapeutic target in BL. Finally, the antitumoral effects of MYC inhibition were further enhanced by BCL6 degradation, suggesting that targeting of MYC and BCL6 could represent an efficacious therapeutic approach in BL.
Methods
Cell lines and reagents
BL cell lines JD38, NAMALWA, RAJI, DAUDI, and RAMOS were purchased from DSMZ (German Collection of Microorganism and Cell Cultures) and the American Type Culture Collection (Manassas, VA). Cells were aliquoted at early passages and used within 10 to 20 passages from thawing. All lymphoma cell lines were cultured in RPMI 1640 medium (Thermo Fisher Scientific) supplemented with 10% fetal bovine serum (FBS), penicillin-streptomycin (100 U/mL), and l-glutamine (2 mM). All cell lines tested negative for Mycoplasma contamination.
HEK-293T and 293 Phoenix packaging cells were obtained from DSMZ and maintained in Dulbecco’s modified Eagle medium, 10% FBS, penicillin-streptomycin (100 U/mL), and l-glutamine (2 mM).
Murine lymphoma cell lines were obtained from Eμ-myc transgenic mice with the corresponding genotype. Briefly, at the humane end point, mice were euthanized, and tumoral lymph nodes were resected. Single-cell suspensions were prepared from fresh tumoral lymph nodes using mechanical disaggregation and isolated with a 40-μm nylon cell strainer (BD Biosystems). Cells were grown in RPMI 1640 supplemented with 15% FBS, penicillin-streptomycin (100 U/mL), and l-glutamine (2 mM) and cultured for ≥4 weeks before proceeding with further experiments. All cell lines were grown at 37°C in a humidified atmosphere with 5% CO2. Cell lines with stable Omomyc expression or FBXO11 deletion/depletion were generated by lentiviral transduction and puromycin selection (as detailed in “Virus preparation and cell transduction”). Where indicated, the following drugs were used for the indicated times and concentrations: cycloheximide (Sigma), doxycycline hyclate (Sigma), puromycin (Santa Cruz Biotechnology, Dallas, TX), blasticidin (Calbiochem), doxorubicin (Selleckchem), and FX1 (Selleckchem). BI-3812 and BI-3802 were obtained by opnMe, Boehringer Ingelheim.
Generation of FBXO11 conditional mice
C57BL/6 mouse embryonic stem cells heterozygous for a targeted allele of FBXO11 were obtained fromthe International Knockout Mouse Consortium (EUCOMM). Embryonic stem cells were microinjected into blastocysts derived from C57BL/6 mice after removal of the LacZ-neomycin cassette by flippase-mediated recombination. The resulting FBXO11 conditional allele has 2 loxP sites inserted into intronic regions upstream and downstream of exon 4 that allow disruption of the FBXO11 sequence after Cre recombination.
FBXO11fl/+ mice were bred with CD19-Cretg/+ transgenic mice (F1 generation) to specifically delete FBXO11 in CD19+ B cells. Mice from the F1 generation were intercrossed to generate cohorts of FBXO11fl/fl/CD19-Cretg/+ (KO), FBXO11fl/+/CD19-Cretg/+ (HET), and FBXO11+/+/CD19-Cretg/+ (WT) littermates.
Eμ-myc [B6.Cg-Tg(IghMyc)22Bri/J; #002728] mice were obtained from The Jackson Laboratory. CD19-Cretg/+ [B6.129P2(C)-Cd19tm1(cre)Cgn/J; stock number 006785] transgenic mice were obtained from F.W. Alt (Harvard Medical School). All mice were housed and maintained in the specific pathogen–free facility at Boston Children’s Hospital. Animal experiments were performed under protocols approved by the Institutional Animal Care and Use Committee of Boston Children’s Hospital (Protocol 16-01-3093R) or by the Italian Ministry of Health for the University of Torino (approval no. 2542017-PR). None of the mice were excluded from the analysis, and no randomization or blinding method was used.
In vivo xenograft experiments
NOD scid γ (NSG) immunocompromised mice were purchased from Charles River Laboratories. For subcutaneous xenografts, RAJI cell lines (FBXO11 WT or KO), either transduced with inducible lentiviral vectors expressing Omomyc complimentary DNA or not, were injected in both flanks of NSG mice; 15 × 106 cells were treated with BI-3802 (5 μM), resuspended in 150 μL of PBS, and injected. Injected mice were administered drinking water containing doxycycline (1 mg/mL; Sigma) to express Omomyc complementary DNA (cDNA). Tumor growth was measured with a caliper every 2 days. Mice were euthanized at the human end point.
Virus preparation and cell transduction
Lentiviruses were produced using the third-generation production system. Briefly, HEK293T cells were maintained in 10% FBS-containing Dulbecco’s modified Eagle medium. Cells at 70% confluence were cotransfected with pVSVG, pCMVR8.74, pRSV-Rev, and a lentiviral vector expressing the construct of interest. Medium was replenished 12 hours after transfection, and the supernatant was collected after 24 and 48 hours. Collected supernatants were passed through a 0.45-μm filter, pooled, and used either fresh or snap frozen. For infection, 4 × 105 cells were infected with the prepared lentivirus along with Polybrene (8 μg/mL), and cells with viral particles were spun down at 2400 rpm for 90 minutes and incubated at 37°C overnight. Target cell selection was begun at 48 hours posttransduction.
Construction of the lentiviral vector pTRIPZ-Omomy-RFP for the inducible expression of an Omomyc-RFP fusion protein was described previously.25 Human and murine lymphoma cell lines were infected with the virus for 24 hours and then selected with puromycin. For cell-sorting enrichment, cells were induced with 1 μg/mL doxycycline for 36 hours and sorted for RFP expression on a FACSAria Sorter (BD Pharmingen). Retroviruses were generated by transfection of pWZL Blast vector expressing CRE-ERT2 in 293 Phoenix packaging cells. Transfected cells were incubated at 37°C for 12 or 18 hours, and supernatants containing viral particles were collected at 24 and 48 hours. Three hundred microliters of retroviral supernatants was used to transduce 5 × 104 lymphoma cells, as previously described.26 CRE-ERT2–transduced cells were selected using blasticidin (Calbiochem) at 25 μg/mL for 6 days.
FBXO11 KO by CRISPR/Cas9 gene editing
FBXO11-KO BL cell lines were generated using CRISPR/Cas9-mediated gene targeting. Guides targeting exon 4 of FBXO11 (sgRNAs #1 and #2) (5'-CCTGCTGAACAGTATCTTC-3', 5'-CTTCTCTTACTTGCTGGAAC-3') were designed using the CRISPR design tool (http://crispr.mit.edu) and ligated into the LentiCRISPR v2 vector (Addgene plasmid #52961).
Lentiviral particles were produced in HEK293T cells and viral supernatant used to transduce BL cell lines (RAJI and DAUDI), according to the protocol described above. Puromycin was added after 48 hours, using the minimal toxic dose per cell line to select transduced cells. The selected cells were seeded as single colonies in 96-well plates by serial dilutions. Clonal cells from individual wells were screened by western blotting and genomic sequencing of the sgRNA target region to identify cell lines that are FBXO11 deficient. DNA fragments covering the sgRNA target region were amplified using the primers int3F (5'-GTGCAAGAGTGTCTGGAAAA-3') and int3R (5'-AATACCACCTGGACTGACAG-3'). To confirm the biallelic deletion of the clones (biallelic KO), the polymerase chain reaction (PCR) products were cloned and sequenced.
Lymphoma cases/tumor samples and mutational analysis
Follicular lymphoma (FL; n = 100), marginal zone lymphoma (MZL; n = 50), and BL (n = 24) tumor samples were obtained from the archives of the Department of Pathology, University of Torino. Ten additional BL samples were from the Department of Pathology, University of Verona (Italy). All patients provided informed written consent. All cases were diagnosed according to international guidelines and patients provided consent according to internal protocols. Details about sample collection for each patient are summarized in supplemental Table 9.
Mutational analysis/DNA extraction, amplification, and sequencing
Genomic DNA was isolated using QIAamp DNA kits (QIAGEN). The complete coding sequences and exon/intron junctions of FBXO11 were analyzed by PCR amplification and classic Sanger sequencing, as previously described.6
Classical Sanger sequencing was outsourced to a reliable service provider (BaseClear, BV, Leiden, The Netherlands). Sequences were compared with the corresponding germline sequences or the reference sequences using the MacVector Version 12.7 software package (http://www.macvector.com). The reference sequences for all annotated exons and flanking introns of FBXO11 were obtained from the UCSC Human Genome database using the messenger RNA (mRNA) accession NM_001190274.1. Synonymous mutations, previously reported polymorphisms (Human dbSNP Database at National Center for Biotechnology Information, build 130, and Ensembl database), and changes present in the matched normal DNA were excluded. For FFPE FL sample #93, the DNA was extracted from the 2 tumoral components (high grade and low grade) using a QIAamp DNA FFPE kit (QIAGEN), following manufacturer’s instructions. PCR for exon 18 of FBXO11 was performed using the following primers: 17F-5'-TCTAGGCTGTATAGAAGACAATGAAA-3' and 17R-5'-GCCACCATCTCTTCCATCAT-3'. PCR products were subjected to Sanger sequencing on both strands, as described above.
The presence of TP53 gene variants was evaluated according to the European Research Initiative on Chronic Lymphocytic Leukemia guidelines.27 Briefly, genomic DNA extracted from the samples was amplified for the identification of the TP53 entire coding region (exons 2-11) following the International Agency for Research on Cancer primers and protocols (http://p53.iarc.fr/ProtocolsAndTools.aspx), with the exception that a unique touchdown PCR cycle was used for all of the sets of primers based on our laboratory experience. PCR conditions consisted of an annealing decrease of 3°C for each of 3 cycles starting from 64°C and down to 57°C, repeated for 44 cycles. Amplicons were purified and run, after direct Sanger sequencing with the same sets of primers used for the PCR, on an AB3130 capillary sequencer (Applied Biosystems, Foster City, CA). Alignment of sequences with the TP53 reference sequence (NC_000017.11), variant description at the cDNA and protein levels, and variant frequency percentage were performed with Mutation Surveyor v2.02 software program; variants were analyzed using the International Agency for Research on Cancer TP53, Seshat, Cosmic, and GLASS Web-based tools to discriminate neutral polymorphisms and benign and pathological variants. An independent round of PCR and sequencing was performed to confirm the variants identified.
FBXO11 functional studies
Functional studies of the FBXO11 activity of various mutants were performed as previously described.6 Full-length FBXO11 transcript variant 1 (NM_025133.4), cloned with an in-frame N terminal FLAG (and HA tag) in the pLVX-IRES-mCherry vector, was kindly provided by Michele Pagano. FBXO11 variants were generated by site-directed mutagenesis using a QuikChange II site-directed mutagenesis kit (Agilent Technologies) and the oligonucleotides 5'-GCGTCTAATTTTGGGGTTGTTTCCAGTCCTTATCTGAAC-3' and 5'-GTTCAGATAAGGACTGGAAACAACCCCAAAATTAGACGC-3' for the S672N mutant, 5'-ACAGATAGTAATCCTACACTAAGAAATAAAATCCATGATGGAAGAG-3' and 5'-CTCTTCCATCATGGATTTTATTTCTTAGTGTAGGATTACTATCTGT-3' for the R723del form, 5'-GGTGTTCTCATCAGCACTAAGTCATCCAATCTTAAGGAA-3' and 5'-TTCCTTAAGATTGGATGACTTAGTGCTGATGAGAACACC-3' for the N763fs*12 form, 5'-AAGCCTCCAAACCGTTTGTTAAAAATCTGATTGCCTTCTAG-3' and 5'-CTAGAAGGCAATCAGATTTTTAACAAACGGTTTGGAGGCTT-3' for the N799K variant and 5'-TGTCATAAAAATAAACACCAAACTGCTTGCCACTGTGTATCC-3' and 5'-GGATACACAGTGGCAAGCAGTTTGGTGTTTATTTTTATGACA-3' for the V641F variant.
FBXO11 splice site variants were generated by amplification of the aberrant transcripts from the cDNA of the mutated samples and cloning in the pLVX-IRES-mCherry vector using BamHI-BamHI sites. All constructs were sequenced and validated before experimental use. FLAG-tagged SNAIL (clone ID: 16218) and FLAG-tagged BCL6 (clone ID: 31391) plasmids were purchased from Addgene (Cambridge, MA).
Cycloheximide pulse-chase assay
HEK293T cells were transfected with the wild-type (WT) or mutated FBXO11 plasmids using a calcium phosphate kit (Clontech Laboratories), following the manufacturer’s instructions. Cycloheximide (CHX) pulse-chase experiments were performed as described before.6 HEK293T cells were seeded on a 6-well plate at a density of 2 to 4 × 105 cells per well. The morning after, the cells were transfected with different plasmids (3 μg total). Twenty-four hours after transfection, the cells were treated with 100 μg/mL CHX dissolved in dimethyl sulfoxide (DMSO); total protein lysate was collected at different time points and subjected to immunoblotting for FBXO11 protein, BCL6, or SNAIL and related protein loading controls.
For CHX pulse-chase experiments in FBXO11 null-RAJI cell lines and Eμ-myc–derived lymphoma cell lines, a total of 3 × 106 cells was plated into 6-well plates, at a concentration of 1 × 106 cells per milliliter. CHX (100 μg/mL for human BL or 100 ng/mL for mouse lymphoma lines) was added to culture medium, and the cells were harvested at the indicated times thereafter.
Reverse transcriptase PCR and quantitative real-time PCR
Total RNA was isolated from cells using TRIzol solution (Life Technologies) and an RNase-free DNase I set (Roche) was used for genomic DNA digestion. cDNA was transcribed using an iScript cDNA synthesis kit (Bio-Rad Laboratories), following the manufacturer’s instructions. For the analysis of FBXO11 aberrant transcripts from BL samples, PCR amplification was performed using primers annealing to the regions corresponding to exons 12 to 17 (12F-5'-CCCTACAGTGGTTCGATGTGA-3'; 17R-5'-TCTGTCCTCCCCAGATTTTG-3'), exons 17 to 20 (17F-5'-TCTAGGCTGTATAGAAGACAATGAAA-3'; 20R-5'-CCTCCAAACCGGTTGTTAAA-3'), and exons 12 to 20 (12F-5'-CCCTACAGTGGTTCGATGTGA-3'; 20R-5'-CCTCCAAACCGGTTGTTAAA-3'). The resulting PCR products were resolved in a 2% agarose gel, and visible bands were excised and purified using a PureLink Quick Gel Extraction kit (Life Technologies). Products were sequenced as described above. In BL case #6, carrying multiple genetic variants, the reverse transcriptase PCR products (exons 12-20) were cloned and sequenced to span both events and determine the allelic distribution of the mutations.
For mouse TP53 mutation analysis, the DNA binding domain of TP53 (hotspot region for mutations) was amplified from primary tumors of mice (FBXO11 WT or KO) using primers binding to exons 4 to 9 (supplemental Table 10). Mutations were identified by Sanger sequencing.
All quantitative reverse transcriptase PCR experiments were performed in triplicate on an ICycler iQ Real-Time PCR Detection System using SYBR Green Supermix (both from Bio-Rad Laboratories). Expression levels for individual transcripts were normalized against the housekeeping genes Gapdh for murine samples or HPRT for human samples. Relative expression was calculated using 2-ΔΔCt (change in cycling threshold) method. Primer pairs are listed in supplemental Table 10.
Protein extraction and immunoblot analysis
Immunoblot analysis was performed by standard methodology. Briefly, GST-FISH buffer (10 mM MgCl2, 150 mM NaCl, 1% NP-40, 2% glycerol, 1 mM EDTA, 25 mM HEPES [pH 7.5]) supplemented with protease inhibitors (Roche), 1 mM phenylmethanesulfonyl fluoride, 10 mM NaF, and 1 mM Na3VO4 was used to isolate proteins from cells or tissues. Protein samples were heat denatured and equally loaded after quantification (Bio-Rad Laboratories protein assay) on sodium dodecyl sulfate–polyacrylamide gels (Bio-Rad Laboratories), run under reducing conditions, and transferred to nitrocellulose (GE Healthcare). Membranes were incubated with specific antibodies, detected with peroxidase-conjugated secondary antibodies (GE Healthcare), and enhanced using a chemiluminescent reagent (Amersham). We used the following primary antibodies: anti–β-actin (Sigma, #A2066), anti-FBXO11 (Sigma, HPA002690), anti-FBXO11 (Bethyl Laboratories, A-301-178A), anti-FLAG (Sigma, #F1804, clone M2), anti-human BCL6 (Dako, #M7211, clone PG-B6p), anti-mouse BCL6 (Cell Signaling Technology, D65C10,), anti-SNAIL (Cell Signaling Technology, #3895, L70G2), and anti-DTL/CDT2 (Bethyl Laboratories, A300-948A).
BCL6 and SNAIL protein abundance was measured using ImageJ software and normalized for the GFP and β-actin intensity of the corresponding lane, respectively.
Histology, immunohistochemistry, and immunofluorescence
For histology, tissue samples were fixed in formalin and embedded in paraffin, cut into 4-μm-thick sections, and stained with hematoxylin and eosin (H&E). For immunohistochemistry, formalin-fixed sections were dewaxed in xylene and dehydrated by passage through graded alcohols to water; sections were microwaved in citrate buffer, pH 6, for 15 minutes and then transferred to phosphate-buffered saline (PBS). Endogenous peroxidase was blocked using 1.6% H2O2 in PBS for 10 minutes, followed by washing in distilled water. Normal serum diluted to 10% in 1% bovine serum albumin was used to block nonspecific staining. Next, the slides were incubated for 1 hour with the following primary antibodies: anti-cleaved caspase 3 (Cell Signaling Technology, clone 5A1E), anti-murine Ki-67 (Abcam, #ab16667, clone SP6), and anti-human BCL6 (Dako, #M7211, clone PG-B6p). After washing, sections were incubated with biotinylated secondary goat antibody to rabbit immunoglobulin G (IgG) and visualized with the EnVision system (Dako).
For immunofluorescence, formalin-fixed sections were dewaxed and rehydrated before performing antigen retrieval with citrate buffer (pH 6). Sections were permeabilized with 0.05% Tween-20, incubated with Protein block solution (Dako) for 30 minutes to block nonspecific staining and then with the primary antibodies against Ki-67 (Dako, #M7240, clone MIB-1) and Omomyc (rabbit polyclonal, from the laboratory of L.S.) overnight at 4°C. Samples were stained with fluorescent secondary antibodies (Alexa Fluor 488 goat anti-mouse or Alexa Fluor 594 goat anti-rabbit; Invitrogen) for 1 hour. Coverslips were mounted on microscope slides using Abcam DAPI mounting medium and left to dry for ≥24 hours. Images were taken with a Nikon epifluorescence microscope using the 20× objective.
Flow cytometry
Single-cell suspensions were prepared from fresh tumors and spleen by mechanical dissociation and were isolated using 40-μm nylon cell strainers (BD Biosystems). Cells were resuspended in PBS and stained with antibodies for 15 minutes, washed, and resuspended in PBS. Data were acquired on a BD FACSVerse flow cytometer (BD Bioscience) and analyzed using FlowJo software (TreeStar).
Flow cytometry analysis of freshly isolated cells from tumors and spleen of Eμ-myc and FBXO11fl/fl/CD19-Cretg/+ transgenic mice was performed using the following fluorescent-labeled anti-mouse antibodies: APC-conjugated anti-mouse CD45R/B220 (BD Biosciences, #561880, clone RA3-6B2) and APC-conjugated anti-CD19 (Miltenyi Biotec, #130-123-791, clone 6D5); PE-conjugated anti-IgG1 (BD Biosciences, #550083, clone A85-1) and APC-conjugated anti-CD4 (clone GK1.5; Miltenyi Biotec #130-116-546); APC/Cy7-conjugated anti-IgM (BioLegend, #406515, clone RMM-1) and APC-conjugated anti-CD3e (Invitrogen, #47-0033-82, clone 500A2); and PE-conjugated anti-GL7 (Invitrogen, #12-5902-82; clone GL-7), and PE-conjugated anti-CD8a (Miltenyi Biotec #130-102-595, clone 53-6.7).
Cell viability and cell proliferation assays
A cell viability assay was performed using CellTiter-Glo (Promega), according to the manufacturer’s instructions. Briefly, cells were seeded into white-walled 96-well plates (3 wells per sample) in the presence or absence of treatment. CellTiter-Glo reagent was added to each well, and luminescence output data were taken at 0, 24, 48, and 72 hours using a GloMax-Multi Detection System (Promega). FX1 (Selleckchem, #S8591) was diluted in DMSO and used at the indicated concentrations. Alternatively, cell viability was assessed using a colorimetric MTS assay (CellTiter 96 AQueous Non-Radioactive Cell Proliferation Assay [MTS] Powder; Promega).
For long-term proliferation assays, cells were seeded at a density of 1 x 105 cells per milliliter in 24-wells plate. Compounds (doxycycline, Omomyc mini-protein, BI-3802, or BI-3812) were added, and every 2 or 3 days cells were counted and split to 1 x 105 cells per milliliter. Upon splitting, fresh compound was added to keep the concentration constant. Cell number and viability were determined by dye exclusion Trypan blue assay using Cell Countess (Life Technologies). Split rates were multiplied to derive proliferation factors. BI-3802 and BI-3812 were kindly provided by opnMe (Boehringer Ingelheim), diluted in DMSO, and used at the indicated concentrations. Omomyc mini-protein was described previously.28
RNA sequencing data analysis
Total RNA was isolated from BL cell lines using TRIzol solution (Life Technologies) and treated with a RNase-free DNase I set (Roche) to digest genomic DNA. RNA sample quality was assessed with an Agilent 2100 Bioanalyzer. RNA concentration was estimated by Qubit. Sequencing libraries were prepared starting with 100 ng of total RNA using a TruSeq RNA Library Prep Kit v2 (Illumina), following the manufacturer’s instructions. The qualities of the libraries were assessed with an Agilent 2100 Bioanalyzer. Sequencing libraries were loaded on a NextSeq 500 Illumina sequencer to generate 75-nt paired-end reads. Fastq files were produced via Illumina bcl2fastq2 (v 2.17.1.14) starting from .bcl files produced by Illumina NextSeq sequencer. Fastq files were analyzed using docker4seq framework.29,30 Specifically, the overall quality of the data was estimated via multiQC.31 Gene level expression was computed using STAR (v 2.5.0)/RSEM (v 1.3.0) software,32,33 using ENSEMBL GRCh38 human genome as reference. Differential expression analysis was estimated via DESeq2,34 also embedded in docker4seq framework. Genes were considered differentially expressed and retained for further analysis with |log2FoldChange| ≥ 1 and a false discovery rate ≤ 0.1.
The following Gene Set Enrichment Analysis data sets were used for RNA sequencing data analysis: ARACNE_TARGET, ARACNe-predicted BCL6 network35 ; M0, genes detected to contain the M0 motif35 ; INR, genes characterized by the Inr motifs, most likely reflecting the presence of BCL6 bound to Miz-135 ; M00424-M2-M0, genes containing M00424-M2-M0 DNA motives present in BCL6 bound promoters as detected by combining chromatin immunoprecipitation with glass slide DNA microarrays (ChIP-on-chip)35 ; CHIP_2_CHIP_CI2009, BCL6 target genes detected in primary centroblasts and DLBCL cells35,36 using ChIP-on-chip; and DW_CI2009, BCL6 target genes detected in primary centroblasts and DLBCL cells by transcription analysis.35,36
Patients from the ICGC MMML-Seq cohort with BL_solid diagnosis were divided into patients who showed a mutation in FBXO11 (8 patients) and patients who did not (12 patients). The 5 normal GC B-cell data sets from the consortium served as control. Mapping and counting were performed as described elsewhere.23 Differential expression was evaluated using DESeq237 in R for the mutated patients vs control and for the unmutated patients vs control separately. For further analysis, genes that showed an absolute log fold change > 1 and an adjusted P value < .1 were used. This corresponded to 3836 differentially expressed genes for patients who exhibited FBXO11 mutations, and 3763 differentially expressed genes for patients without FBXO11 mutation. The genes were sorted with respect to their expression fold change, and preranked Gene Set Enrichment Analysis38,39 was performed on the public server of Genepattern.org using the BCL6 hallmarks, a minimum set size of 5, No Collapse, 1000 permutations, and weighted enrichment statistics.
Statistical analysis
Statistical analysis was performed with GraphPad Prism 7 software. P values were calculated using an unpaired 2-tailed Student t test with Welch’s correction, as indicated in each figure legend. Kaplan-Meier survival curves were created with GraphPad Prism 7, and P values were determined using a log-rank (Mantel-Cox) test.
Results
FBXO11 is frequently inactivated in BL
To comprehensively describe FBXO11 alterations in BL, we sequenced a new cohort of BL cases, as well as curated FBXO11 mutations in larger published cohorts of sporadic and nonsporadic BL cases.22,23 We first sequenced a new series of BCL6+ lymphomas that included 100 cases of FL, 34 cases of BL, and 7 BL cell lines. We also sequenced 50 cases of MZL, because a previous report found recurrent mutations of FBXO11 in splenic MZL.40 In this new series of cases, nonsynonymous mutations and deletions of FBXO11 were identified in 1 of 100 FL cases (1%), 1 of 50 MZL cases (2%), 7 of 34 BL cases (20.6%), and 3 of 7 BL cell lines (43%), for a total of 10 of 41 (24.4%) mutated BLs (supplemental Figure 1A). Sequencing of available paired normal DNA in patients with BL showed that FBXO11 mutations are somatic, as we described previously for DLBCL6 (supplemental Figure 1B). In the 2 mutated FL and MZL cases, an area with transformation to high-grade DLBCL lymphoma was present. When we dissected and sequenced the low-grade FL or MZL lymphoma and the associated high-grade transformed lymphoma separately, the FBXO11 mutation was only detected in the high-grade component of the lymphoma (supplemental Figure 1C-D), indicating that, in these 2 cases, FBXO11 mutations were acquired late, possibly during lymphoma transformation. This is in keeping with previous data showing that BCL6 alterations in MALT lymphoma are typically found in the associated large B-cell lymphoma component.41 An extended analysis of 88 additional FL cases confirmed that FBXO11 mutations are rare in FL, with 2 mutated cases, 1 of which was associated again with a DLBCL component (Figure 1A; supplemental Figure 1E).
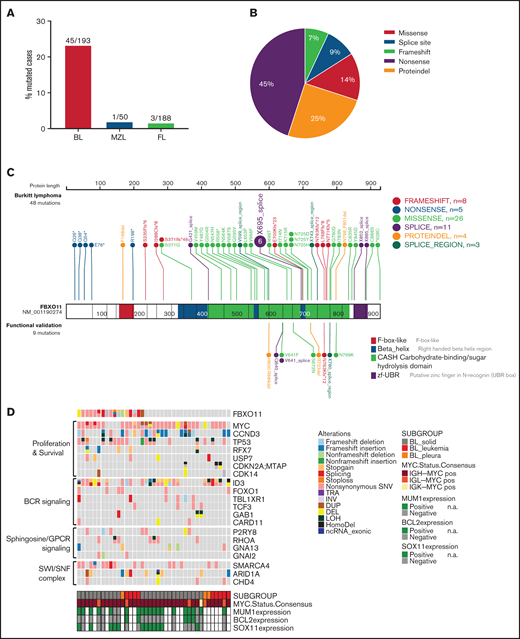
FBXO11 is frequently mutated in BL. (A) Bar graph showing the frequency of FBXO11 nonsynonymous mutations in B-cell lymphomas: BL, MZL, and FL. (B) Distribution of FBXO11 genetic alterations in BL. (C) Lollipop plot displaying 57 nonsynonymous FBXO11 mutations identified in BL primary cases (n = 193). Mutations that were functionally validated are shown below the gene model. (D) Mutation status of BL-associated genes in 39 BL cases.23 The affected genes are reordered in pathways to highlight any mutual exclusivity. Mutations are colored according to the type of SNVs, SVs, and CNAs. Cases are displayed according to the BL subgroups and are annotated with MUM1, BCL2, and SOX11 expression status analyzed by immunohistochemistry, as well as MYC translocation status. BCR, B-cell receptor; CNAs, copy number alterations; DEL, deletion; DUP, duplication; HomoDel, homozygous deletion; INV, inversion; LOH, loss of heterozygosity; n.a., not available; ncRNA, non-coding RNA; SNVs, single nucleotide variants; SVs, structural variants; TRA, translocation.
FBXO11 is frequently mutated in BL. (A) Bar graph showing the frequency of FBXO11 nonsynonymous mutations in B-cell lymphomas: BL, MZL, and FL. (B) Distribution of FBXO11 genetic alterations in BL. (C) Lollipop plot displaying 57 nonsynonymous FBXO11 mutations identified in BL primary cases (n = 193). Mutations that were functionally validated are shown below the gene model. (D) Mutation status of BL-associated genes in 39 BL cases.23 The affected genes are reordered in pathways to highlight any mutual exclusivity. Mutations are colored according to the type of SNVs, SVs, and CNAs. Cases are displayed according to the BL subgroups and are annotated with MUM1, BCL2, and SOX11 expression status analyzed by immunohistochemistry, as well as MYC translocation status. BCR, B-cell receptor; CNAs, copy number alterations; DEL, deletion; DUP, duplication; HomoDel, homozygous deletion; INV, inversion; LOH, loss of heterozygosity; n.a., not available; ncRNA, non-coding RNA; SNVs, single nucleotide variants; SVs, structural variants; TRA, translocation.
Next, we reviewed FBXO11 mutations in published larger cohorts of sporadic and nonsporadic BL cases.22,23 FBXO11 single nucleotide variants (SNVs) and small indels (insertions and deletions) were confirmed in 38 of 159 cases, and larger structural variants (large deletions, inversions and duplications) were confirmed in 5 of 159 cases. In total, we found FBXO11 mutations (SNVs and indels) in 45 of 193 BL cases (23%), with 10 cases (5%) harboring ≥2 FBXO11 mutations, suggesting a biallelic FBXO11 inactivation. Missense mutations were the most common, followed by splice site mutations (Figure 1A-C; supplemental Tables 1 and 2). Frameshift or non-sense mutations clustered in early exons, making them likely to cause a complete loss of protein expression. In contrast, missense mutations clustered in the 3 CASH domains and in the terminal zinc finger domain that are important for FBXO11 ubiquitination function (Figure 1C). The CASH domains in FBXO11 (aa 418-837) are critical for the interaction with BCL6,6 whereas the interaction site for SKP1 to form the substrate-specificity module of the E3 ubiquitin ligase42 is likely within the F-box like domain (aa 153-201). In an additional published series, FBXO11 variants were identified in 37 of 101 BL cases, of which 7 were missense mutations and 1 was a frameshift mutation.24 Overall, these data corroborate that FBXO11 is one of the most frequently mutated genes in BL. The distribution of the FBXO11 mutations was similar to those described in DLBCL (supplemental Figure 1F)
In our new series, we were able to validate some splicing mutations by mRNA sequencing. In case #4, a nucleotide change was found within the consensus splice donor site at the exon 15–intron 15 junction, resulting in the presence of an aberrant FBXO11 mRNA with in-frame loss of exon 15. In case #6, a donor splice site mutation at the exon 19–intron 19 junction led to skipping of the entire exon 19. In case #7, the first base of exon 16 was mutated (c.1921G>T), causing an amino acid change (V641F) and skipping of the entire exon 16. The loss of exon 16 caused an out-of-frame translation that generated a premature stop codon and a truncated polypeptide (supplemental Figure 2A-D). Sequencing analysis of the FBXO11 transcripts in case #6 that showed a FBXO11 mutation and a genomic deletion encompassing exons 15/16 demonstrated that the splice site mutation and the deletion were on independent alleles, thereby leading to biallelic inactivation of the gene (supplemental Figure 2E-G).
When correlating FBXO11 mutations with other genes frequently mutated in BL, we found a significant positive correlation between FBXO11 mutations and p53 mutations (p53MUT/FBXO11MUT = 52%, p53MUT/FBXO11WT = 33%, n = 193, P = .0185) and with SMARCA4 mutations (n = 159, P = .0088), whereas no correlation was found between FBXO11 mutations and any other recurrently mutated gene in BL, including ID3, DDX3X, CCND3, ARID1A, or other genes associated with tonic B-cell receptor signaling or cell cycle regulation (Figure 1D; supplemental Figure 2H; supplemental Tables 3 and 4). By analyzing the variant allelic frequency (VAF) from 80× whole-genome sequencing data, we found that, in most cases, the VAF of p53MUT was higher than that of FBXO11MUT, indicating that p53 mutations may precede FBXO11 mutations in BL during clonal evolution more often than not (supplemental Table 5).
Biochemical and biological validation of FBXO11 loss-of-function mutations
To determine the biological activity of FBXO11 mutations, we generated several mutant constructs for recurrent mutations. BCL6 and SNAIL are the 2 most important targets of FBXO11; therefore, their stability was analyzed to determine FBXO11 activity in HEK-293T cells expressing the WT or mutant constructs. As we described previously,6 overexpression of the WT FBXO11 protein accelerated BCL6 and SNAIL degradation; in contrast, all tumor-derived mutants displayed an impaired ability to induce BCL6 (Figure 2A,C) and SNAIL degradation (Figure 2B,D), thus demonstrating that all of the analyzed mutations generate a loss-of-function FBXO11 protein. In the N799K mutant, BCL6 and SNAIL degradation appeared comparable to WT. However, the stability of the FBXO11N799K protein itself was reduced compared with WT and all of the other mutants, suggesting that this mutation could impair FBXO11 activity indirectly by reducing protein abundance rather than by directly affecting the enzymatic activity. Destabilization of the FBXO11 protein has also been suggested as a potential mechanism for the loss of function secondary to FBXO11 mutations in neurodevelopmental disorders.15
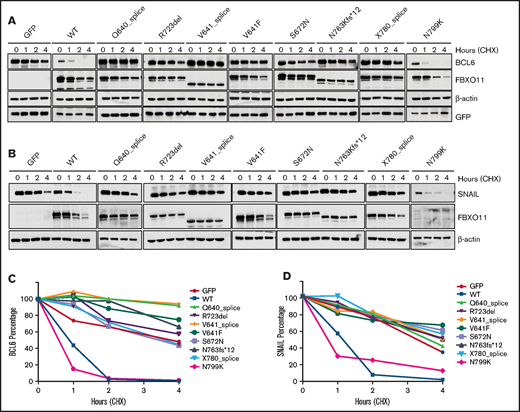
FBXO11 tumor-derived mutants show impaired ability to induce BCL6 and SNAIL degradation. (A) Representative western blot for FBXO11 and BCL6 protein expression in HEK-293T cells transfected with BCL6 in combination with GFP, WT FBXO11, or FBXO11 mutants at the indicated time points after treatment with CHX. FBXO11 mutants were detected by anti-FBXO11 antibody, and BCL6 was detected by anti-BCL6 antibody. β-actin was used as a loading control. GFP was used for transfection efficiency. One representative experiment of 2 independently performed experiments is shown. (B) Impaired degradation of SNAIL by FBXO11 mutants. Representative western blot for FBXO11 and SNAIL protein expression in HEK-293T cells transfected with SNAIL and WT FBXO11 or the indicated FBXO11 mutants at the indicated time points after treatment with CHX. FBXO11 mutants were detected by anti-Flag antibody. β-actin was used as a loading control. One representative experiment of 2 independently performed experiments is shown. (C) Degradation kinetics of BCL6 by FBXO11 mutants. BCL6 abundance was measured from western blot gels as in (A) by ImageJ software and normalized for the GFP intensity of the corresponding lane. The ratio between the relative levels of BCL6/GFP at each time 0 was set as 100%. Data are from 1 of 2 independent experiments, each with comparable results. (D) SNAIL abundance was measured from western blot gels, as in (B), using ImageJ software and normalized for β-actin intensity of the corresponding lane. The ratio between the relative levels of SNAIL/β-actin at each time 0 was set as 100%. Data are from 1 of 2 independent experiments, each with comparable results.
FBXO11 tumor-derived mutants show impaired ability to induce BCL6 and SNAIL degradation. (A) Representative western blot for FBXO11 and BCL6 protein expression in HEK-293T cells transfected with BCL6 in combination with GFP, WT FBXO11, or FBXO11 mutants at the indicated time points after treatment with CHX. FBXO11 mutants were detected by anti-FBXO11 antibody, and BCL6 was detected by anti-BCL6 antibody. β-actin was used as a loading control. GFP was used for transfection efficiency. One representative experiment of 2 independently performed experiments is shown. (B) Impaired degradation of SNAIL by FBXO11 mutants. Representative western blot for FBXO11 and SNAIL protein expression in HEK-293T cells transfected with SNAIL and WT FBXO11 or the indicated FBXO11 mutants at the indicated time points after treatment with CHX. FBXO11 mutants were detected by anti-Flag antibody. β-actin was used as a loading control. One representative experiment of 2 independently performed experiments is shown. (C) Degradation kinetics of BCL6 by FBXO11 mutants. BCL6 abundance was measured from western blot gels as in (A) by ImageJ software and normalized for the GFP intensity of the corresponding lane. The ratio between the relative levels of BCL6/GFP at each time 0 was set as 100%. Data are from 1 of 2 independent experiments, each with comparable results. (D) SNAIL abundance was measured from western blot gels, as in (B), using ImageJ software and normalized for β-actin intensity of the corresponding lane. The ratio between the relative levels of SNAIL/β-actin at each time 0 was set as 100%. Data are from 1 of 2 independent experiments, each with comparable results.
To investigate the biological role of loss of FBXO11 activity, we generated a mouse model with conditional inactivation of FBXO11 by designing a conditional KO FBXO11fl allele with LoxP sequences flanking exon 4 of the FBXO11 gene (supplemental Figure 3A). We crossed FBXO11fl/fl mice with CD19-Cre mice to selectively delete the FBXO11 allele in B cells. Splenocytes isolated from CD19-Cre/FBXO11+/+, CD19-Cre/FBXO11fl/+, and CD19-Cre/FBXO11fl/fl mice showed the effective deletion of exon 4, resulting in reduced FBXO11 mRNA in FBXO11fl/fl mice, likely as a result of non-sense–mediated decay (supplemental Figure 3B-C). Consistently, FBXO11 protein was reduced in CD19-Cre/FBXO11fl/+ splenocytes and was undetectable in CD19-Cre/FBXO11fl/fl splenocytes (supplemental Figure 3D). CD19-Cre/FBXO11fl/+ and CD19-Cre/FBXO11fl/fl mice survived for up to 250 days and did not develop lymphoma without significant alterations in the B-cell populations, including total B cells, IgM+ and switched IgG1+ B cells, or GC B cells (B220+/GL7+) (supplemental Figure 3E), consistent with a previous study.20
Because deregulation of MYC expression is a hallmark of BL, we investigated whether loss of 1 or 2 copies of FBXO11 cooperated with MYC-driven lymphomagenesis. Eμ-myc mice have primarily been used to study MYC-driven lymphomagenesis, including cooperation of MYC with loss of function of ID3, another gene that is frequently mutated in BL.24 All Eμ-myc mice died of lymphoma within 200 days, with a time course of tumor development similar to other studies.43,44 Remarkably, lymphoma onset was accelerated significantly and survival was shortened in Eμ-myc/CD19-Cre/FBXO11fl/+ and Eμ-myc/CD19-Cre/FBXO11fl/fl mice (Figure 3A). In keeping with previous studies,43,44 Eμ-myc mice developed lymphoma at a very similar frequency, exhibiting a phenotype of immature (B220+/IgM−) or more mature (B220+/IgM+) B cells; in contrast, Eμ-myc/CD19-Cre/FBXO11fl/+ and Eμ-myc/CD19-Cre/FBXO11fl/fl mice primarily developed B220+/IgM+/IgD+ lymphoma (Figure 3B-D), implicating FBXO11 in the rapidity of lymphoma development, as well as in the maturation of lymphoma cells. This is reminiscent of a skew toward cluster 2 in the classification of Eμ-myc lymphoma, which includes lymphoma with a more follicular/marginal zone and GC B-cell stage differentiation.45 Systemic spread in organs, including spleen, lymph nodes, bone marrow, kidney, liver, and lungs, occurred at the same frequency in all genotypes (supplemental Table 6).
![FBXO11 deletion accelerates lymphomagenesis in Eμ-myc–transgenic mice. (A) Kaplan-Meier survival analysis of Eμ-myc (n = 33), Eμ-myc/FBXO11fl/fl;CD19-Cretg/+ (KO) (n = 21) and Eμ-myc/FBXO11fl/+;CD19-Cretg/+ (HET) (n = 16) mice, with a median survival time of 121, 89, and 85 days, respectively. Eμ-myc (n = 33) are a combination of Eμ-myc/FBXO11fl/fl (n = 23) and Eμ-myc/FBXO11fl/+ (n = 10). P < .0001 log-rank (Mantel-Cox) test. (B) B220 and IgM expression in lymphomas from Eμ-myc (n = 17), Eμ-myc/FBXO11fl/fl;CD19-Cretg/+ (KO) (n = 16), and Eμ-myc/FBXO11fl/+;CD19-Cretg/+ (HET) (n = 9) mice. (C) B220 and IgD expression in lymphomas from Eμ-myc (n = 16), Eμ-myc/FBXO11fl/fl;CD19-Cretg/+ (KO) (n = 10), and Eμ-myc/FBXO11fl/+;CD19-Cretg/+ (HET) (n = 5) mice. (D) Representative flow cytometry of B220 and IgM expression in lymphoma from Eμ-myc, Eμ-myc/FBXO11-KO, and Eμ-myc/FBXO11 HET mice. (E) Representative H&E stains (top row) and immunohistochemistry for cleaved caspase 3 (middle row) and Ki-67 (bottom row) performed on Eμ-myc primary lymphoma with the indicated genotypes (original magnification ×200). Scale bars, 50 μm. Insets show high-magnification images. (F) Quantification of macrophages in Eμ-myc lymphoma with the indicated genotypes (n = 6 mice for each genotype). Data are mean (± standard deviation [SD]) macrophages per 40× field. Quantification of cleaved caspase 3+ (G) and Ki-67+ (H) cells in Eμ-myc lymphoma with the indicated genotypes (n = 4 mice for each genotype). Data are mean ± SD. (I) BCL6 stability in Eμ-myc/FBXO11-KO lymphoma cells. Western blot showing BCL6 protein expression after treatment with CHX for the indicated times in 3 immortalized Eμ-myc/FBXO11fl/fl lymphoma cell lines transduced with a tamoxifen-inducible CreERT2 vector. Cre recombinase was activated by treatment with 10 nM 4-hydroxytamoxifen (4-OHT) for 4 hours to induce deletion of the FBXO11 floxed gene. A20 is a mouse BCL6+ B lymphoma cell line used as control. **P < .01, ***P < .001, unpaired 2-tailed Student t test.](https://ash.silverchair-cdn.com/ash/content_public/journal/bloodadvances/5/23/10.1182_bloodadvances.2021005682/3/m_advancesadv2021005682f3.png?Expires=1765913453&Signature=ZlYL0h~eflGPm4xxLVIEPEK2T9dQg6bHVU4~gQCCl4fTv07Z0DZyNxOSdRhMUEA34bS5ybEn5A1TAXjh4Ca4-h6cFkTZaadwLX~oKqZ0A4E5kqA2c20bAQUj4VYFqt2vrQLxOzYiEUuNKKgvU016EyzRy9VoUDpvN4zF0MZOBrUQkO~xV~FP7sH9LiQRUfQRIo~jIIbZjTBdPsllaVpWUX6Ihrb4Pq4Vbdj8Q6FPLnz6zJkykWkdsgh53gWb-njwJlFAHM9T-uG3xcl2IJlszw-~an6oB2Ohl~SH1uaYmFRdgOszC1YdOd4~FbAOQx7nmdov4odrjIEgbLpviCySlQ__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
FBXO11 deletion accelerates lymphomagenesis in Eμ-myc–transgenic mice. (A) Kaplan-Meier survival analysis of Eμ-myc (n = 33), Eμ-myc/FBXO11fl/fl;CD19-Cretg/+ (KO) (n = 21) and Eμ-myc/FBXO11fl/+;CD19-Cretg/+ (HET) (n = 16) mice, with a median survival time of 121, 89, and 85 days, respectively. Eμ-myc (n = 33) are a combination of Eμ-myc/FBXO11fl/fl (n = 23) and Eμ-myc/FBXO11fl/+ (n = 10). P < .0001 log-rank (Mantel-Cox) test. (B) B220 and IgM expression in lymphomas from Eμ-myc (n = 17), Eμ-myc/FBXO11fl/fl;CD19-Cretg/+ (KO) (n = 16), and Eμ-myc/FBXO11fl/+;CD19-Cretg/+ (HET) (n = 9) mice. (C) B220 and IgD expression in lymphomas from Eμ-myc (n = 16), Eμ-myc/FBXO11fl/fl;CD19-Cretg/+ (KO) (n = 10), and Eμ-myc/FBXO11fl/+;CD19-Cretg/+ (HET) (n = 5) mice. (D) Representative flow cytometry of B220 and IgM expression in lymphoma from Eμ-myc, Eμ-myc/FBXO11-KO, and Eμ-myc/FBXO11 HET mice. (E) Representative H&E stains (top row) and immunohistochemistry for cleaved caspase 3 (middle row) and Ki-67 (bottom row) performed on Eμ-myc primary lymphoma with the indicated genotypes (original magnification ×200). Scale bars, 50 μm. Insets show high-magnification images. (F) Quantification of macrophages in Eμ-myc lymphoma with the indicated genotypes (n = 6 mice for each genotype). Data are mean (± standard deviation [SD]) macrophages per 40× field. Quantification of cleaved caspase 3+ (G) and Ki-67+ (H) cells in Eμ-myc lymphoma with the indicated genotypes (n = 4 mice for each genotype). Data are mean ± SD. (I) BCL6 stability in Eμ-myc/FBXO11-KO lymphoma cells. Western blot showing BCL6 protein expression after treatment with CHX for the indicated times in 3 immortalized Eμ-myc/FBXO11fl/fl lymphoma cell lines transduced with a tamoxifen-inducible CreERT2 vector. Cre recombinase was activated by treatment with 10 nM 4-hydroxytamoxifen (4-OHT) for 4 hours to induce deletion of the FBXO11 floxed gene. A20 is a mouse BCL6+ B lymphoma cell line used as control. **P < .01, ***P < .001, unpaired 2-tailed Student t test.
FBXO11 deletion accelerates lymphomagenesis in Eμ-myc–transgenic mice. (A) Kaplan-Meier survival analysis of Eμ-myc (n = 33), Eμ-myc/FBXO11fl/fl;CD19-Cretg/+ (KO) (n = 21) and Eμ-myc/FBXO11fl/+;CD19-Cretg/+ (HET) (n = 16) mice, with a median survival time of 121, 89, and 85 days, respectively. Eμ-myc (n = 33) are a combination of Eμ-myc/FBXO11fl/fl (n = 23) and Eμ-myc/FBXO11fl/+ (n = 10). P < .0001 log-rank (Mantel-Cox) test. (B) B220 and IgM expression in lymphomas from Eμ-myc (n = 17), Eμ-myc/FBXO11fl/fl;CD19-Cretg/+ (KO) (n = 16), and Eμ-myc/FBXO11fl/+;CD19-Cretg/+ (HET) (n = 9) mice. (C) B220 and IgD expression in lymphomas from Eμ-myc (n = 16), Eμ-myc/FBXO11fl/fl;CD19-Cretg/+ (KO) (n = 10), and Eμ-myc/FBXO11fl/+;CD19-Cretg/+ (HET) (n = 5) mice. (D) Representative flow cytometry of B220 and IgM expression in lymphoma from Eμ-myc, Eμ-myc/FBXO11-KO, and Eμ-myc/FBXO11 HET mice. (E) Representative H&E stains (top row) and immunohistochemistry for cleaved caspase 3 (middle row) and Ki-67 (bottom row) performed on Eμ-myc primary lymphoma with the indicated genotypes (original magnification ×200). Scale bars, 50 μm. Insets show high-magnification images. (F) Quantification of macrophages in Eμ-myc lymphoma with the indicated genotypes (n = 6 mice for each genotype). Data are mean (± standard deviation [SD]) macrophages per 40× field. Quantification of cleaved caspase 3+ (G) and Ki-67+ (H) cells in Eμ-myc lymphoma with the indicated genotypes (n = 4 mice for each genotype). Data are mean ± SD. (I) BCL6 stability in Eμ-myc/FBXO11-KO lymphoma cells. Western blot showing BCL6 protein expression after treatment with CHX for the indicated times in 3 immortalized Eμ-myc/FBXO11fl/fl lymphoma cell lines transduced with a tamoxifen-inducible CreERT2 vector. Cre recombinase was activated by treatment with 10 nM 4-hydroxytamoxifen (4-OHT) for 4 hours to induce deletion of the FBXO11 floxed gene. A20 is a mouse BCL6+ B lymphoma cell line used as control. **P < .01, ***P < .001, unpaired 2-tailed Student t test.
In human BL lymphoma, the overexpression of MYC induces a potent drive toward proliferation associated with activation of the apoptotic program.46 As a result, BL typically shows a high fraction of proliferating cells associated with a high number of macrophages clearing apoptotic cells (so-called “starry sky” appearance).47 Compared with Eμ-myc mice, Eμ-myc/CD19-Cre/FBXO11fl/fl mice showed a reduced number of intratumoral macrophages and decreased apoptosis, whereas proliferation was equally high in all genetic backgrounds (Figure 3E-H); this indicates that deletion of FBXO11 contributes to the acceleration of MYC-driven lymphomagenesis by decreasing apoptosis rather than increasing cell proliferation, which is already extremely high in BL.47
FBXO11 deletion increases BCL6 stability in MYC-driven mouse lymphoma and human BL
RNA sequencing performed in Eμ-myc lymphoma indicated that, among the known substrates of FBXO11, CDT2/DTL is expressed at the highest levels, BCL6 is expressed at lower levels, and SNAIL and BLIMP-1 are minimally expressed (supplemental Figure 4A). Despite being low in most Eμ-myc lymphomas,48 expression of BCL6 protein was relatively increased in Eμ-myc/CD19-Cre/FBXO11fl/fl lymphomas (supplemental Figure 4B-C), without changes in mRNA levels (supplemental Figure 4D), consistent with protein stabilization in FBXO11-deficient lymphoma. To study the effects of FBXO11 deletion in Eμ-myc lymphoma more directly, we immortalized several cell lines from Eμ-myc/FBXO11fl/fl mice and transduced them with a vector expressing a tamoxifen-inducible Cre recombinase. In Eμ-myc/FBXO11fl/fl lines, the addition of tamoxifen resulted in the rapid loss of FBXO11 protein expression that was accompanied by stabilization and an increased abundance of BCL6 protein (Figure 3I), indicating that BCL6 degradation is also regulated by FBXO11 in Eμ-myc lymphoma. Because p53 is believed to be regulated by BCL649 and is frequently inactivated in Eμ-myc lymphoma,50 we next sequenced TP53 in a series of Eμ-myc and Eμ-myc/CD19-Cre/FBXO11fl/fl lymphomas. Although Eμ-myc lymphoma showed the expected frequency of mutations (6/12 = 50%) consistent with protein stabilization (supplemental Table 7), the frequency of TP53 mutations in Eμ-myc/CD19-Cre/FBXO11fl/fl lymphomas was significantly lower (1/16 = 6%; P = .0228). Overall, these data indicate that deletion of FBXO11 in MYC-driven lymphoma in mice stabilizes BCL6 and obviates the need for a TP53 mutation, possibly as a result of repression of the p53 pathway as a consequence of BCL6 stabilization.
We next examined whether FBXO11 deletion affected BCL6 stability in human BL cells. BL typically expresses abundant BCL6 in most cells (supplemental Figure 5A). We generated FBXO11-KO BL cells by disrupting the endogenous FBXO11 gene in 2 BL cell lines (RAJI and Daudi) via a CRISPR/Cas9 approach, with 2 sgRNAs targeting exon 4 of the FBXO11 gene (supplemental Figure 5B-E). Remarkably, in all FBXO11-KO lines, the BCL6 protein was completely stabilized at similar levels to the FARAGE cell line, a DLBCL line with homozygous deletion of the FBXO11 gene6 (Figure 4A-C), indicating that FBXO11 is a necessary ubiquitin ligase for control of BCL6 levels in BL. We also evaluated other substrates of FBXO11 in BL. BLIMP-1 and SNAIL were not expressed at detectable levels in BL lines, consistent with previous findings51 (supplemental Figure 5F). CDT2/DTL was expressed, but its protein levels were not significantly stabilized by FBXO11 deletion (supplemental Figure 5G-H).
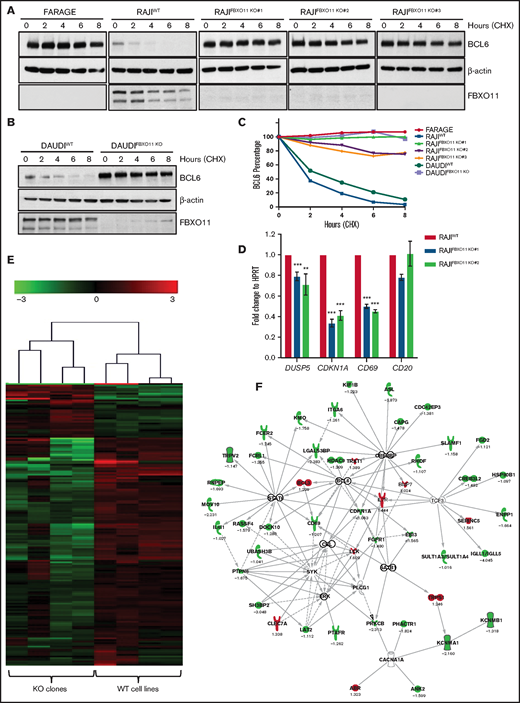
FBXO11 deletion in BL cell lines leads to BCL6 protein stabilization. (A) BLC6 stability in human BL lines in which FBXO11 is knocked out. Representative western blot showing BCL6 protein expression in 3 independent FBXO11 KO RAJI cell lines (clone #1 obtained with sgRNA#1, clones #2 and #3 obtained with sgRNA#2; supplemental Figure 5B-E) after treatment with CHX for the indicated times. FARAGE cell line, which is homozygous for deletion of the FBXO11 gene, was used as control for BCL6 stability. One representative experiment of 3 independently performed experiments is shown. (B) Representative western blot showing BCL6 protein expression in WT or FBXO11-KO DAUDI cell line after treatment with CHX for the indicated times. β-actin was used as a loading control. One representative experiment of 3 independently performed experiments is shown. (C) Degradation kinetics of BCL6 in BL WT or FBXO11-KO cells. BCL6 abundance was measured from western blot gels as in (A-B) using ImageJ software and normalized for the β-actin intensity of the corresponding lane. (D) Quantitative real-time PCR expression analysis of the BCL6 target genes DUSP5, CDKN1A, and CD69 mRNA in RAJI and 2 FBXO11-KO RAJI cell lines (#1 and #2). Data are mean ± standard deviation. (E) Hierarchical clustering (Euclidean distance, average linkage) of 172 genes detected as differentially expressed between the FBXO11-WT or -KO RAJI cell lines. (F) Analysis of genes detected as differentially expressed between WT cell lines and KO clones. Forty-seven of 172 differentially expressed genes can be connected to each other using Ingenuity Pathway Analysis (QIAGEN IPA) in a BCL6-centered network. **P < .01, ***P < .001, unpaired 2-tailed Student t test.
FBXO11 deletion in BL cell lines leads to BCL6 protein stabilization. (A) BLC6 stability in human BL lines in which FBXO11 is knocked out. Representative western blot showing BCL6 protein expression in 3 independent FBXO11 KO RAJI cell lines (clone #1 obtained with sgRNA#1, clones #2 and #3 obtained with sgRNA#2; supplemental Figure 5B-E) after treatment with CHX for the indicated times. FARAGE cell line, which is homozygous for deletion of the FBXO11 gene, was used as control for BCL6 stability. One representative experiment of 3 independently performed experiments is shown. (B) Representative western blot showing BCL6 protein expression in WT or FBXO11-KO DAUDI cell line after treatment with CHX for the indicated times. β-actin was used as a loading control. One representative experiment of 3 independently performed experiments is shown. (C) Degradation kinetics of BCL6 in BL WT or FBXO11-KO cells. BCL6 abundance was measured from western blot gels as in (A-B) using ImageJ software and normalized for the β-actin intensity of the corresponding lane. (D) Quantitative real-time PCR expression analysis of the BCL6 target genes DUSP5, CDKN1A, and CD69 mRNA in RAJI and 2 FBXO11-KO RAJI cell lines (#1 and #2). Data are mean ± standard deviation. (E) Hierarchical clustering (Euclidean distance, average linkage) of 172 genes detected as differentially expressed between the FBXO11-WT or -KO RAJI cell lines. (F) Analysis of genes detected as differentially expressed between WT cell lines and KO clones. Forty-seven of 172 differentially expressed genes can be connected to each other using Ingenuity Pathway Analysis (QIAGEN IPA) in a BCL6-centered network. **P < .01, ***P < .001, unpaired 2-tailed Student t test.
BCL6 is a transcriptional repressor that has multiple targets1 that may be responsible for lymphoma. Consistently, stabilization of BCL6 in FBXO11-KO BL cell lines resulted in the expected transcriptional changes of its known targets, such as decreased expression of BCL6 transcriptional targets DUSP5, CDKN1A, and CD69 (Figure 4D). Comparison of RNA sequencing profiles from WT and KO human lines showed a set of 172 genes modified upon deletion of FBXO11 (Figure 4E; supplemental Table 8). Connectivity networks generated by Ingenuity Pathway Analysis of the differentially expressed genes identified at the top of a pathway linked to BCL6 (Figure 4F). We performed gene set enrichment analysis (GSEA) against an extensive database of BCL6-correlated gene sets.35 We obtained significant enrichment for DW_CI2009 (family-wise error rate [FWER], P = .003), ARACNE_TARGET (FWER P = .057), and M0 (FWER P = .1) associated with KO cell lines (supplemental Figure 6A). Furthermore, running GSEA against human gene sets v7.1 (h.all.v7.1.symbols), FBXO11-KO cells showed enrichment in the Wnt (FWER P = .009), interferon-α (FWER P < .001), and interferon-γ (FWER P < .001) pathways, which have been shown to be associated with BCL635 (supplemental Figure 6B). We also performed GSEA analysis for BCL6 hallmark genes that were differentially expressed between control GC B cells and FBXO11-mutated solid BL cases (n = 8) or FBXO11 WT solid BL cases (n = 12). FBXO11 WT and mutated cases showed significant enrichment of the BCL6 signature compared with normal GC B cells (supplemental Figure 6C).
Targeted degradation of BCL6 combined with MYC inhibition is a powerful therapeutic approach for BL
BL is currently treated by chemotherapy; no targeted therapy has been approved, in contrast to other mature B-cell lymphomas.52 Because we showed that the effects of FBXO11 deletions are mainly mediated by BCL6 stabilization, we reasoned that BCL6 could be a valuable therapeutic target in BL. Recently, small molecules have been developed that efficiently block BCL6 transcriptional activity, such as FX153 and BI-3812,54 or target BCL6 for degradation, such as BI-3802.54 BI-3802 induces polymerization of BCL6 coupled to highly specific protein degradation, resulting in increased pharmacological activity compared with other BCL6 inhibitors.55 The FX1 inhibitor and the BI-3802 degrader efficiently derepressed BCL6 transcriptional targets in WT and FBXO11-KO cells (supplemental Figure 7A-B). The BCL6 degrader BI-3802, but not the BI-3812 inhibitor, induced a rapid and almost complete degradation of BCL6 in WT cells, as well as in FBXO11-KO cells, despite their higher basal levels of BCL6 (Figure 5A). Degradation of BCL6 was observed to last for ≥6 days upon drug removal and extensive washing (supplemental Figure 7C). Similarly, in mouse Eμ-myc lymphoma cells, BI-3802, but not BI-3812, induced rapid and durable BCL6 degradation (Figure 5B; supplemental Figure 7D). Treatment with the BCL6 degrader BI-3802 or with FX1 or BI-3812 inhibitors significantly reduced cell growth in vitro of human WT or FBXO11-KO BL cells (Figure 5C; supplemental Figure 8A-B) and mouse Eμ-myc and Eμ-myc/FBXO11-KO cell lines (supplemental Figure 8C-D). BL tumor xenografts in which BCL6 degradation was induced by pretreatment with BI-3802 grew significantly slower than did control tumors in mice (Figure 5D). Therefore, targeting BCL6 with inhibitors or degraders resulted in decreased growth of human BL and mouse MYC-driven lymphomas.
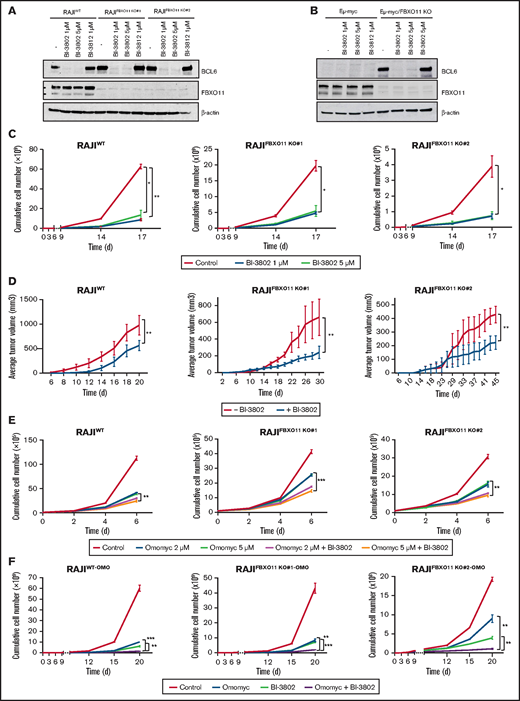
BCL6 degradation impairs BL growth and potentiates MYC inhibition. (A) Western blot analyses for BCL6 expression on the indicated BL cell lines. Cells were treated for 60 minutes with BI-3802 (1 or 5 μM), BI-3812 (1 μM), or vehicle (-) before collection. Data are representative of 2 experiments. β-actin was used as loading control. (B) Western blot analysis for BCL6 on immortalized lymphoma cell lines obtained from FBXO11-WT (Eμ-myc) or -KO (Eμ-myc/FBXO11fl/fl;CD19-Cretg/+) mice. Cells were treated for 1 hour with BI-3802 (1 or 5 μM), BI-3812 (1 μM), or vehicle (-) before collection. Data are representative of 2 experiments with similar results. β-actin was used as loading control. (C) Effects of BCL6 degradation on WT and FBXO11-KO RAJI (clones #1 and #2) cell growth. Cells were kept at constant concentrations of the degrader BI-3802 as indicated (1 μM or 5 μM) and split to 100 000 cells per milliliter every 3 days. Split rates were multiplied to derive growth curves. Data are mean ± standard error of the mean (SEM) and are representative of ≥3 independent experiments. *P < .05, **P < .01, unpaired 2-tailed Student t test. (D) Growth of tumor xenografts in NSG mice of WT and FBXO11-KO RAJI (clones #1 and #2) cells treated with BI-3802 (5 μM) for 24 hours before injection. Data (n = 5 mice per group) are shown as mean ± SEM. **P < .01, paired 2-tailed Student t test. (E) Growth curves of WT and FBXO11-KO RAJI (clones #1 and #2) cells treated with Omomyc mini-protein (2 or 5 μM), alone or in combination with BI-3802 (5 μM). Total cell number was quantified at the indicated time points. Data are mean ± SEM of triplicates from a single experiment representative of 2 repeats. **P < .01, ***P < .001, unpaired 2-tailed Student t test. (F) Growth curves of WT and FBXO11-KO RAJI (clones #1 and #2) cells infected with a doxycycline-inducible lentiviral vector expressing Omomyc. Cells were treated only with doxycycline to induce Omomyc expression (Omomyc), or BI-3802 (5 μM) or a combination of doxycycline and BI-3802 (Omomyc + BI-3802). Total cell number was quantified at the indicated time points. Data are mean ± SEM of triplicates from a single experiment representative of two repeats. **P < .01, ***P < .001, unpaired 2-tailed Student t test.
BCL6 degradation impairs BL growth and potentiates MYC inhibition. (A) Western blot analyses for BCL6 expression on the indicated BL cell lines. Cells were treated for 60 minutes with BI-3802 (1 or 5 μM), BI-3812 (1 μM), or vehicle (-) before collection. Data are representative of 2 experiments. β-actin was used as loading control. (B) Western blot analysis for BCL6 on immortalized lymphoma cell lines obtained from FBXO11-WT (Eμ-myc) or -KO (Eμ-myc/FBXO11fl/fl;CD19-Cretg/+) mice. Cells were treated for 1 hour with BI-3802 (1 or 5 μM), BI-3812 (1 μM), or vehicle (-) before collection. Data are representative of 2 experiments with similar results. β-actin was used as loading control. (C) Effects of BCL6 degradation on WT and FBXO11-KO RAJI (clones #1 and #2) cell growth. Cells were kept at constant concentrations of the degrader BI-3802 as indicated (1 μM or 5 μM) and split to 100 000 cells per milliliter every 3 days. Split rates were multiplied to derive growth curves. Data are mean ± standard error of the mean (SEM) and are representative of ≥3 independent experiments. *P < .05, **P < .01, unpaired 2-tailed Student t test. (D) Growth of tumor xenografts in NSG mice of WT and FBXO11-KO RAJI (clones #1 and #2) cells treated with BI-3802 (5 μM) for 24 hours before injection. Data (n = 5 mice per group) are shown as mean ± SEM. **P < .01, paired 2-tailed Student t test. (E) Growth curves of WT and FBXO11-KO RAJI (clones #1 and #2) cells treated with Omomyc mini-protein (2 or 5 μM), alone or in combination with BI-3802 (5 μM). Total cell number was quantified at the indicated time points. Data are mean ± SEM of triplicates from a single experiment representative of 2 repeats. **P < .01, ***P < .001, unpaired 2-tailed Student t test. (F) Growth curves of WT and FBXO11-KO RAJI (clones #1 and #2) cells infected with a doxycycline-inducible lentiviral vector expressing Omomyc. Cells were treated only with doxycycline to induce Omomyc expression (Omomyc), or BI-3802 (5 μM) or a combination of doxycycline and BI-3802 (Omomyc + BI-3802). Total cell number was quantified at the indicated time points. Data are mean ± SEM of triplicates from a single experiment representative of two repeats. **P < .01, ***P < .001, unpaired 2-tailed Student t test.
Drugs that specifically target MYC activity in BL are not clinically available. Bromodomain and extra-terminal repeat (BET) inhibitors have been proposed to treat MYC-driven lymphoma, as the best approximation of a MYC inhibition; however they have not been approved for clinical use.56 In contrast, we explored a more specific MYC targeting achieved via a dominant-negative mini-protein.57 A cell-permeable Omomyc mini-protein effectively interferes with MYC transcriptional activity, impairing cancer cell growth in various models with an 50% inhibitory concentration ranging from ∼6 μM to 26 μM in different cell lines.28 Treatment of WT or FBXO11-KO BL cells with 20 μM or 10 μM Omomyc completely suppressed cell growth and markedly increased apoptosis of BL cells (supplemental Figure 9). When cells were treated with lower doses of Omomyc (5 μM or 2 μM), the effects on cell growth were reduced, but combination with the BCL6 degrader BI-3802 further potentiated the cytostatic effect (Figure 5E). Next, we generated mouse and human WT and FBXO11-KO cell lines that express a doxycycline-inducible Omomyc28 (supplemental Figure 10A-B). Induction of Omomyc expression was sufficient to reduce the growth of FBXO11-WT and -KO cells with an effect comparable to cells treated with lower concentrations of Omomyc; the combination with BI-3802 further reduced cell growth in human and mouse lines (Figure 5F; supplemental Figure 10C-D). Finally, we evaluated the effects of MYC blockade via Omomyc in combination with BCL6 degradation in vivo. Omomyc expression was induced by doxycycline administration to mice bearing BL xenografts pretreated with BI-3802. The Omomyc and BI-3802 combination impaired tumor growth significantly more than did Omomyc or BI-3082 alone (Figure 6A; supplemental Figure 10E). The impairment of tumor growth was due to increased apoptosis and suppression of cell proliferation (Figure 6B-D). Remarkably, cells expressing Omomyc were counterselected in vivo, and the residual cells with Omomyc expression showed a reduced proliferation rate (Figure 6E-F). Overall, these data support dual targeting of MYC and BCL6 as an effective therapeutic approach in BL.
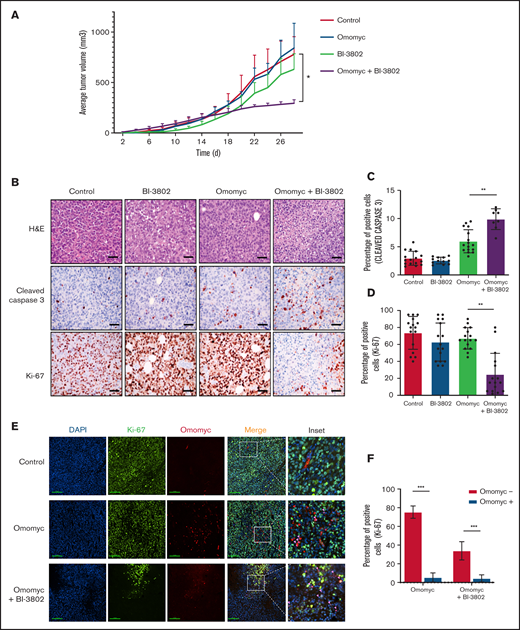
Combined targeting of MYC and BCL6 severely impairs BL growth in mice by reducing proliferation and increasing apoptosis. (A) Growth of tumor xenografts of RAJI cells transduced with doxycycline-inducible Omomyc vector in NSG mice. Lymphoma cells were treated with BI-3802 (5 μM) for 24 hours before injection. Mice were administered normal water (red line) or doxycycline water (blue line) from day 0. Data are mean ± standard error of the mean of 8 tumors per group. *P < .05, paired 2-tailed Student t test. (B) Representative H&E stains (top row) and immunohistochemistry for cleaved caspase 3 (middle row) and Ki-67 (bottom row) performed on RAJI lymphoma arising in NSG mice and treated with the BCL6 degrader BI-3802 together with Omomyc, as in (A) (original magnification ×200). Scale bars, 50 μm. Quantification of cleaved caspase 3+ (C) and Ki-67+ (D) cells in RAJI lymphoma with the indicated treatment. Data were obtained from 4 areas in 4 independent tumors for each treatment. Data are mean ± standard deviation. **P < .01, unpaired 2-tailed Student t test. (E) Representative immunofluorescence performed on RAJI lymphoma grafted in NSG mice and treated with the BCL6 degrader BI-3802 together with Omomyc, as in (A). Cells were stained for Ki-67 (green), Omomyc (red), and DAPI (blue) for the nucleus (original magnification, ×200). Scale bars, 100 μm. Higher-magnification images of the white boxes are shown in the far right panels. Pink arrow-heads in inset images indicate Ki-67−/Omomyc+ nuclei. (F) Quantification of the experiment described in (E). Three sections for each treatment were used to score the number of Ki-67+/Omomyc− and Ki-67+/Omomyc+ nuclei. ***P < .001.
Combined targeting of MYC and BCL6 severely impairs BL growth in mice by reducing proliferation and increasing apoptosis. (A) Growth of tumor xenografts of RAJI cells transduced with doxycycline-inducible Omomyc vector in NSG mice. Lymphoma cells were treated with BI-3802 (5 μM) for 24 hours before injection. Mice were administered normal water (red line) or doxycycline water (blue line) from day 0. Data are mean ± standard error of the mean of 8 tumors per group. *P < .05, paired 2-tailed Student t test. (B) Representative H&E stains (top row) and immunohistochemistry for cleaved caspase 3 (middle row) and Ki-67 (bottom row) performed on RAJI lymphoma arising in NSG mice and treated with the BCL6 degrader BI-3802 together with Omomyc, as in (A) (original magnification ×200). Scale bars, 50 μm. Quantification of cleaved caspase 3+ (C) and Ki-67+ (D) cells in RAJI lymphoma with the indicated treatment. Data were obtained from 4 areas in 4 independent tumors for each treatment. Data are mean ± standard deviation. **P < .01, unpaired 2-tailed Student t test. (E) Representative immunofluorescence performed on RAJI lymphoma grafted in NSG mice and treated with the BCL6 degrader BI-3802 together with Omomyc, as in (A). Cells were stained for Ki-67 (green), Omomyc (red), and DAPI (blue) for the nucleus (original magnification, ×200). Scale bars, 100 μm. Higher-magnification images of the white boxes are shown in the far right panels. Pink arrow-heads in inset images indicate Ki-67−/Omomyc+ nuclei. (F) Quantification of the experiment described in (E). Three sections for each treatment were used to score the number of Ki-67+/Omomyc− and Ki-67+/Omomyc+ nuclei. ***P < .001.
Discussion
Our findings establish that FBXO11 is 1 of the most frequently mutated genes in BL. We show that the critical function of FBXO11 in BL is the regulation of BCL6 and, therefore, identify BCL6 as a promising specific therapeutic target in BL. Based on our newly identified sequences and cases and the published data set, we determined that FBXO11 is mutated or deleted in ∼26% of BL cases but very rarely in FL and MZL (1% and 2%, respectively). Thus, frequent FBXO11 mutations appear to be restricted to aggressive types of B-cell lymphoma, including DLBCL and BL. In the 2 FL and MZL cases with mutated FBXO11, the mutation was present only in the high-grade transformed component of the lymphoma, as we showed by sequencing of the microdissected areas. FBXO11 mutations are rare in FL and are associated with transformation to DLBCL, in keeping with the knowledge that dysregulation of BCL6 expression in FL by other genetic events, such as a chromosomal translocation, is associated with increased transformation to DLBCL.58
The frequency of FBXO11 mutations in BL is substantially higher than in DLBCL,6 indicating that BL is likely the B-cell lymphoma with the highest frequency of FBXO11 mutations. Early exon FBXO11 mutations identified in BL were frameshift mutations leading to out-of-frame loss of FBXO11 expression by premature termination or non-sense–mediated mRNA decay, resulting in FBXO11 haploinsufficiency. In contrast, mutations in late exons were more frequently missense mutations clustered in the CASH domains or the C terminus zinc-finger domain (Figure 1B). These domains are critical for the ubiquitin-ligase activity of FBXO11, and mutations arising in these domains generate loss-of-function protein with possible dominant-negative activity.6
The genetic hallmark of BL is the translocation involving the MYC oncogene with the immunoglobulin heavy chain locus, or more rarely, 1 of the immunoglobulin light-chain loci,59,60 where dysregulated MYC expression is driven by the immunoglobulin enhancers.61 In addition to the hallmark MYC translocations, recent sequencing studies have shown recurrent mutations in few other genes in both sporadic and endemic BL, including mutations in TP53, ID3, TCF3, CCND3, DDX3X, ARID1A, and SMARCA4,22,23,62-64 SIN3A, USP7, and CHD8,22 as well as recurrent mutations in RHOA, P2RY8, and GNA13 that disrupt Gα13 signaling.23 Analysis of gene alterations did not show any significant mutual exclusivity of FBXO11 mutations with other genes, indicating that FBXO11 mutations can be functionally tolerated in combination with other mutations that typically regulate proliferation and survival, the SWI/SNF complex, and tonic B-cell receptor signaling in BL22,23 (supplemental Tables 3 and 4). FBXO11 mutations were equally present in sporadic and endemic Epstein-Barr virus–positive BL (supplemental Figure 2H).22 In contrast, we observed an enrichment of cases that harbored both TP53 and FBXO11 mutations. More in-depth characterization of such cases by calculation of the VAF of TP53 and FBXO11 mutations showed that, in most cases, the VAF of TP53 mutations was greater than that of FBXO11 mutations, suggesting that TP53 mutations were acquired at an earlier stage compared with FBXO11 mutations during BL clonal evolution (supplemental Table 5).
Genetic experiments in mice showed that FBXO11 haploinsufficiency accelerates lymphoma onset driven by deregulated expression of MYC. Although Eμ-myc mice are not a perfect model for human BL, they have been extensively characterized and are useful to study the cooperation of genetic alterations with MYC-driven lymphomagenesis.24 Notably, we observed a comparable acceleration in mice with a conditional monoallelic or biallelic loss of FBXO11 in B cells, suggesting that loss of only 1 copy of FBXO11 is sufficient to accelerate lymphoma onset significantly (Figure 3A). This is in keeping with findings in human DLBCL and BL, which typically carry heterozygous FBXO11 mutations or deletions. Monoallelic FBXO11 mutations also caused higher rates of lymphoproliferative disorder than did biallelic FBXO11 deletion in Cγ1-Cre–driven conditional B cells. Also, DLBCL was observed only rarely in FBXO11-haploinsufficient mice after a long latency, indicating the requirement of additional genetic alterations in FBXO11-deficient mice for full lymphoma development.20 Remarkably, lymphomas that developed in Eμ-myc/CD19-Cre/FBXO11fl/+ or Eμ-myc/CD19-Cre/FBXO11fl/fl mice showed a more mature phenotype, with expression of surface IgM in most cases, compared with Eμ-myc lymphomas. FBXO11 deficiency resulted in stabilization of BCL6 in Eμ-myc lymphoma (Figure 3I). Also, Eμ-myc/CD19-Cre/FBXO11fl/+ and Eμ-myc/CD19-Cre/FBXO11fl/fl mice rarely harbored TP53 mutations, which are frequent in Eμ−myc lymphomas and human BL. These data suggest that the molecular alterations imparted by FBXO11 deficiency might bypass the need for TP53 mutations in MYC-driven lymphoma, possibly as a result of the direct control of FBXO11 on p53 functions,9-11 BCL6-mediated p53 repression,49 or the effects of FBXO11 on the p53 downstream mediator p21.7,8 However, BCL6 is expressed at significant levels only in a subset of Eμ-myc lymphoma. Therefore, it is possible that the effects of FBXO11 loss of function are not limited to BCL6 stabilization. Of the other known substrates of FBXO11 ubiquitin ligase activity, SNAIL,12 and BLIMP-113,14 are poorly expressed in BL, whereas CDT27,8 is expressed in Eμ−myc lymphoma and human BL (supplemental Figures 4A and 5F). CDT2 is a DDB1 and CUL4-associated factors protein that controls cell cycle progression. FBXO11-mediated degradation of CDT2 stabilizes p21 and Set8 and regulates the response to transforming growth factor-β.7,8 Thus, increased cell cycle progression and destabilization of p21 and modulation of transforming growth factor-β mediated by increased CDT2 expression65,66 could further contribute to the accelerated lymphomagenesis in FBXO11-deficient lymphoma cells. Finally, we recently showed that FBXO11 can regulate the levels of CD40 and FAS in normal B cells and BL lymphoma cells,21 thus adding additional molecular mechanisms that could contribute to the accelerated MYC-driven development of lymphoma in FBXO11 deficiency. Finally, it is possible that FBXO11 regulates unknown targets that could contribute to the pathogenesis of BL or other lymphomas.
The frequency of FBXO11 mutations in BL indicates that BCL6 is critical for BL, and the BCL6 network was the top network that was perturbated in FBXO11-KO BL cells (Figure 4F). BCL6 is a critical transcriptional regulator in BL,67,68 and suppression of BCL6 function by HDAC inhibitor–mediated acetylation and chromatin modification enhances BET inhibitor effects in B-cell lymphoma cells.69 A recently developed BCL6 degrader54 markedly reduced BCL6 expression in Eμ-myc lymphoma and human BL, induced derepression of known BCL6 targets (supplemental Figure 7B), and impaired cell growth in vitro and lymphoma growth in vivo in FBXO11-WT and -KO cell lines (Figure 5C-D), suggesting that, when clinically available, BCL6 degraders could be useful in BL therapy, regardless of the FBXO11 mutational status. The kinetics of BL inhibition was comparable to the effects of the BCL6 degrader on DLBCL cell lines, with stasis reached at 4 to 7 days.54 Similar effects were obtained with the BCL6 inhibitors FX1 and BI-3812 at higher concentrations (50% inhibitory concentration = 50 μM). Collectively, these data show that targeting BCL6 as monotherapy has limited potential that should be expanded by combination with other therapies in patients with BL.
MYC is an obvious target of cancer therapy, but direct inhibitors have not been found, whereas BET inhibitors have been proposed as surrogate inhibitors of MYC activity.70 One of the most promising agents is the Omomyc mini-protein that impairs MYC activity by acting as a dominant negative via homo- and heterodimerization.43,44 Indeed, when used at a relatively high concentration (10-20 μM), Omomyc dramatically impaired the growth of WT and FBXO11-KO BL cells. At lower concentrations, Omomyc was less active, but combination with a BCL6 degrader further impaired lymphoma growth (Figures 5E-F and 6). These data indicate that, although a strong inhibition of MYC might be sufficient to block BL growth, synchronous inhibition of BCL6 activity could contribute to the therapeutic activity of a less-complete MYC inhibition. Thus, precise targeting of MYC and BCL6 could represent a promising therapeutic tool for the treatment of BL.
Acknowledgments
The authors thank Michele Pagano and Lukas Cermak for providing plasmid constructs, F.W. Alt for providing CD19-Cre transgenic mice, and Nancy Chamberlin for critical review of the manuscript.
This work was supported by National Institutes of Health National Cancer Institute grant R01 CA196703-01; AIRC fellowship-abroad, project code 15135 (C.P.); Fondazione Umberto Veronesi postdoctoral fellowship 2018-2019 (C.P.); AIRC grant MFAG (M.C.) and Bando Giovani Ricercatori 2009-GR 1603126 (M.C.); AIRC grant IG-23146 (C.V.); and the German Ministry of Science and Education in the framework of the ICGC MMML-Seq (01KU1002), ICGC DE-Mining (01KU1505), and MMML-MYC-SYS (036166B) projects (R.S.).
The visual abstract was drawn with BioRender.
Authorship
Contributions: C.P., T.-C.C., M.C., M.A., R.D., F.L., T.P., E.P., and C.V. performed research, supervised experiments, and analyzed data; Q.W., M.O., and R.A.C. performed bioinformatics analysis; L.B. performed TP53 mutational analysis on human samples; L.M., P.F.d.C., and A.Z. provided clinical samples; C.L., S.H.B., R.S., B.M.G., R.D.M., and L.M.S. provided mutational and clinical data on BL samples; S.M.-M. performed and analyzed immunofluorescence and supervised experiments; J.R.W. and L.S. provided Omomyc plasmid and Omomyc mini-protein and supervised experiments; and C.P. and R.C. conceived the project, designed research, analyzed data, and wrote the manuscript.
Conflict-of-interest disclosure: L.S. is a founder of Peptomyc S.L. L.S. and J.R.W. own shares in Peptomyc S.L. The remaining authors declare no competing financial interests.
Correspondence: Roberto Chiarle, Department of Pathology, Children’s Hospital Boston and Harvard Medical School, Enders 1116.1, 320 Longwood Ave, Boston, MA 02115, e-mail: roberto.chiarle@childrens.harvard.edu.

