

Abstract
Juvenile myelomonocytic leukemia is an overlapping myeloproliferative and myelodysplastic disorder of early childhood . It is associated with a spectrum of diverse outcomes ranging from spontaneous resolution in rare patients to transformation to acute myeloid leukemia in others that is generally fatal. This unpredictable clinical course, along with initially descriptive diagnostic criteria, led to decades of productive international research. Next-generation sequencing now permits more accurate molecular diagnoses in nearly all patients. However, curative treatment is still reliant on allogeneic hematopoietic cell transplantation for most patients, and additional advances will be required to improve risk stratification algorithms that distinguish those that can be observed expectantly from others who require swift hematopoietic cell transplantation.
Diagnosis
Clinical presentation and diagnostic workup
Juvenile myelomonocytic leukemia (JMML) is a rare and frequently fatal myeloproliferative neoplasm of early childhood with an estimated incidence of 1.2 cases per million and a median age at disease onset of ~ 2 years.1 Patients typically present with fever, splenomegaly, thrombocytopenia, and a high circulating white blood cell (WBC) count with peripheral monocytosis and a leukoerythroblastic blood smear. Cough, bloody stools, an erythematous rash, or failure to thrive may also be observed. These symptoms are likely related to infiltration of monocytes into the lungs, gastrointestinal tract, skin, and spleen, respectively.2,3 Elevated hemoglobin F corrected for age is found in ~ 50% of patients. Apart from an extended infectious disease workup, further traditional diagnostic tools include bone marrow aspiration, a peripheral blood smear, and flow cytometry (diagnostic criteria per the 2016 Revision to World Health Organization [WHO] Classification4 are summarized in Table 1). Bone marrow examination can display varying degrees of abnormal myelo- or megakaryopoiesis but must identify fewer than 20% blasts to rule out acute myeloid leukemia (AML). Bone marrow findings typically include hypercellularity with granulocytic hyperplasia and full maturation of myeloid progenitors, although increased monocytes are not always present. Dysplasia can be noted across all 3 hematopoietic lineages, and reticulin fibrosis is occasionally observed. In patients with monosomy 7, a predominance of erythroid precursors can be seen.5–7 Leukoerythroblastosis is not part of the WHO criteria for diagnosis of JMML but is commonly seen on peripheral blood smear and recapitulates the myelodysplastic and myeloproliferative properties of this overlap syndrome. Flow cytometry is often helpful at diagnosis8,9 to rule out other acute leukemias but does not play an important role thereafter unless the clinician is worried about progression to AML. Hyperphosphorylation of STAT5 was previously used as an adjunctive criterion for diagnosis but is not routinely offered in clinical laboratories.10,11 Similarly, hypersensitivity of myeloid progenitor cells to granulocyte-macrophage colony stimulating factor (GM-CSF) in colony-forming assays is a hallmark laboratory feature of JMML.12 However, GM-CSF hypersensitivity can be present in certain viral infections, is not routinely available, is difficult to interpret even in expert hands, and is therefore rarely used to establish a clinical diagnosis of JMML. A molecular confirmation is now possible in nearly every patient with JMML.
Diagnostic criteria for JMML per the 2016 WHO Classification4
| Category I: clinical and hematologic features (all 4 features mandatory) | Category II: genetic studies (1 feature is sufficient) | Category III: other features (patients without features of category II must have ≥2 of the following features) |
|---|---|---|
| Absence of t(9:22) BCR/ABL fusion gene | Somatic mutation in KRAS, NRAS, or PTPN11* | Circulating myeloid or erythroid precursors |
| Absolute monocyte count > 1000/µL | Clinical diagnosis of NF-1 or NF1 gene mutation | Monosomy 7 or other chromosomal abnormality |
| Less than 20% blasts in peripheral blood/bone marrow | Germline CBL mutation or LOH of CBL | WBC > 10 000/μL |
| Splenomegaly | Increased hemoglobin F for age | |
| GM-CSF hypersensitivity† | ||
| Hyperphosphorylation of STAT5† |
| Category I: clinical and hematologic features (all 4 features mandatory) | Category II: genetic studies (1 feature is sufficient) | Category III: other features (patients without features of category II must have ≥2 of the following features) |
|---|---|---|
| Absence of t(9:22) BCR/ABL fusion gene | Somatic mutation in KRAS, NRAS, or PTPN11* | Circulating myeloid or erythroid precursors |
| Absolute monocyte count > 1000/µL | Clinical diagnosis of NF-1 or NF1 gene mutation | Monosomy 7 or other chromosomal abnormality |
| Less than 20% blasts in peripheral blood/bone marrow | Germline CBL mutation or LOH of CBL | WBC > 10 000/μL |
| Splenomegaly | Increased hemoglobin F for age | |
| GM-CSF hypersensitivity† | ||
| Hyperphosphorylation of STAT5† |
Germline mutations need to be excluded.
These tests are not routinely available and are rarely used to make a clinical diagnosis of JMML.
Molecular diagnosis
Nearly all patients with JMML will have mutations detected in the Ras/MAPK signaling pathway, with a small subset of patients harboring translocations or mutations upstream of the Ras pathway. Most mutated genes in JMML can be acquired in the germline or somatic configuration. Our recommendation and practice are therefore to screen every patient suspected of JMML by next-generation sequencing (NGS) platforms, using both a tumor (bone marrow or blood is acceptable) and a normal/germline sample. For the normal sample, we use a buccal swab at diagnosis with the knowledge that these are often contaminated with leukemia cells, but distinguishing germline from somatic origin is still generally possible because a germline lesion will display a variant allele frequency close to 50%. The gold standard for determining the germline status of a mutation is via sequencing of skin fibroblasts. Skin biopsies can be challenging, and if mosaicism is suspected, repeat Sanger sequencing of a buccal swab when there are no circulating monocytes (after treatment) can be considered. Up to 30% of patients will harbor secondary mutations at diagnosis, most of which are subclonal. Of those patients with secondary mutations, up to 10% will have compound mutations in the Ras pathway, with 1 being clonal and 1 being subclonal.13 We therefore sequence patients on a panel capable of returning quantitative allele burdens to distinguish driver from secondary mutations. For rare patients without Ras pathway mutations, we recommend performing RNA sequencing or targeted RNA sequencing to detect potentially targetable fusions. We routinely perform cytogenetics and/or fluorescence in situ hybridization for monosomy 7 and trisomy 8 for all patients at diagnosis. Approximately 30% of patients will have cytogenetic abnormalities, with monosomy 7 being the most common (20% of patients). The likelihood of cytogenetic abnormalities is associated with the JMML genetic subtype, and most frequently, monosomy 7 is observed in KRAS-mutant JMML.3
Genetic subtypes according to driver mutations in the Ras pathway
The biochemical hallmark of JMML is hyperactivation of the Ras/MAPK pathway including RAF/MEK/ERK. The overwhelming majority of patients with JMML (∼95%) will harbor mutations in canonical Ras pathway genes, including NF1, NRAS, KRAS, PTPN11, CBL, RRAS, and RRAS2, with the vast majority of these driver mutations being mutually exclusive.13-15 Germline predisposition (either inherited or de novo) to developing JMML is frequent and occurs in nearly 25% of cases. RASopathies, a group of developmental disorders caused by germline mutations in the Ras/MAPK pathway, have clinical implications later in life and highlight the need for germline testing in patients suspected of having JMML. Patients with neurofibromatosis type 1 and CBL syndrome have germline mutations in NF1 and CBL, respectively, and can develop JMML after acquiring loss of heterozygosity (LOH) in their bone marrow.16–18 In fact, children with neurofibromatosis type 1 (NF1) are at a 300-fold increased risk of JMML or other myeloid malignancies.19 Loss of heterozygosity typically occurs via uniparental isodisomy, where the mutated allele undergoes duplication along with loss of the wild-type allele, maintaining diploidy.18,20 Rarely, patients with mutations in NF1 or CBL undergo LOH through other mechanisms including somatic point mutations, insertions, deletions, or translocations.21,22
Although the mechanism of acquired isodisomy contributing to leukemic development is similar in CBL- and NF1-associated JMML, the clinical course differs significantly. Patients with NF1-associated JMML are often older at diagnosis and present with a higher platelet count, as well as a higher percentage of blasts in their bone marrow compared with patients with JMML with other subtypes.23 In contrast, patients with JMML with germline CBL mutations frequently experience spontaneous disease regression; however, these patients can present with aggressive disease and occasionally require hematopoietic cell transplantation (HCT). Although CBL-related JMML can be indolent, these patients have been noted to develop life-threatening vasculitides later in life, highlighting the diverse and serious nature of manifestations in this syndrome. Interestingly, we recently identified several patients with somatic-only CBL mutations, either heterozygous or homozygous, with no germline CBL mutations. Patients with somatic CBL mutations seem to have a more aggressive phenotype with no cases of spontaneous regression noted to date.24
Noonan syndrome (NS), another genetic disorder with multiple congenital abnormalities, is caused by germline mutations in PTPN11, SOS1, RAF1, KRAS, and NRAS and predisposes for the development of a transient myeloproliferative disease (MPD) of infancy that typically self-resolves but infrequently requires chemotherapy or even HCT in the case of aggressive disease.25,26 No patients with NS-associated MPD have been noted to acquire secondary genetic mutations to date; however, cytogenetic abnormalities, with monosomy 7 in particular, have been reported in rare cases.27,28 Technically, patients with germline mutations in PTPN11, NRAS, or KRAS do not meet the WHO criteria for JMML.4 In contrast, somatic mutations in PTPN11, which are the most common cause of JMML, typically occur at different codons or at similar codons with different amino acid substitutions and lead to stronger phosphatase effects than those affected in NS.29 Similar to PTPN11, germline KRAS mutations in NS have been found to have milder biochemical effects than oncogenic somatic mutations.26
Patients with NRAS-mutated JMML seem to have the greatest clinical diversity among the JMML subtypes, with a considerable percentage of patients relapsing after allogeneic HCT (frequently with co-occurring SETBP1 mutations), whereas others survive with regressing disease in the absence of HCT.1 Mutations in other Ras isoforms, including RRAS and RRAS2, are typically somatic but can also be germline in rare cases.30,31 Finally, mutations in regulators PDE8A, SOS1, and RAC2 have been identified in a few cases.14,15 Rare patients have also been reported to harbor FLT3 tyrosine kinase domain mutations at codons 835 and 836.32,33 Similarly, mutations in SH2B3, in either a germline configuration or somatically acquired, have been observed in patients with JMML as a driver or secondary mutations.13 Clinical characteristics of the 5 main genetic JMML subtypes are described in Table 2.
Genetic, epigenetic, and clinical characteristic of genetic JMML subtypes
| PTPN11 | KRAS | NRAS | NF1 | CBL | |
|---|---|---|---|---|---|
| Prevalence | ∼35-40% | ∼15% | ∼15-20% | ∼10-15% | ∼10-15% |
| Configuration | Germline or somatic | Germline or somatic | Germline or somatic | Germline + LOH | Germline ± LOH or somatic ± LOH |
| Genetic characteristics | Frequent co-occurence with secondary mutations including in NF1 | Frequent association with monosomy 7 | Can co-occur with SETBP1 mutations | Two-thirds of cases have LOH via uniparental disomy | Secondary mutations in additional genes are exceedingly rare |
| Most common DNA methylation subgroup(s) | High | Intermediate | Low | Intermediate or high | Low |
| Germline characteristics | Can present with MPD of infancy | Can present with MPD of infancy | Can present with MPD of infancy | Older age at disease onset Higher platelet count Fatal without HCT High probability of treatment-related mortality | Possibility of spontaneous resolution Indication for HCT is unclear |
| Somatic characteristics | Older age at diagnosis Rapidly fatal without HCT High incidence of relapse after HCT | Heterogenous outcomes | Typically occurs in infants and toddlers Heterogenous outcomes | Somatic-only NF1 mutations are rare but possible | Somatic-only CBL mutations can be associated with a more aggressive disease course |
| PTPN11 | KRAS | NRAS | NF1 | CBL | |
|---|---|---|---|---|---|
| Prevalence | ∼35-40% | ∼15% | ∼15-20% | ∼10-15% | ∼10-15% |
| Configuration | Germline or somatic | Germline or somatic | Germline or somatic | Germline + LOH | Germline ± LOH or somatic ± LOH |
| Genetic characteristics | Frequent co-occurence with secondary mutations including in NF1 | Frequent association with monosomy 7 | Can co-occur with SETBP1 mutations | Two-thirds of cases have LOH via uniparental disomy | Secondary mutations in additional genes are exceedingly rare |
| Most common DNA methylation subgroup(s) | High | Intermediate | Low | Intermediate or high | Low |
| Germline characteristics | Can present with MPD of infancy | Can present with MPD of infancy | Can present with MPD of infancy | Older age at disease onset Higher platelet count Fatal without HCT High probability of treatment-related mortality | Possibility of spontaneous resolution Indication for HCT is unclear |
| Somatic characteristics | Older age at diagnosis Rapidly fatal without HCT High incidence of relapse after HCT | Heterogenous outcomes | Typically occurs in infants and toddlers Heterogenous outcomes | Somatic-only NF1 mutations are rare but possible | Somatic-only CBL mutations can be associated with a more aggressive disease course |
Fusion proteins identified in patients with JMML
Several translocations have been reported in diseases that phenotypically meet criteria of JMML, including most recently in ALK, SOS1, and FLT3, that lead to hyperactivated Ras signaling.15,34 Fusions involving FIP1L1-RARA, HCMOGT-1-PDGFRB, NDEL1-PDGFRB, and NUP98-HOXA11 have also previously been found in patients diagnosed clinically with JMML.35-38 Patients with these fusion proteins are unique in that tyrosine kinase inhibitors or differentiating agents can potentially be used in initial treatment (ie, ALK inhibitors, FLT3 inhibitors, arsenic trioxide, or all-trans-retinoic acid).15,34 In general, the cutoff of 20% bone marrow blasts that differentiates JMML from AML is arbitrary. We are aware of several additional patients with structural rearrangements that are typically associated with AML including inversion 3 and NUP98/NSD1 fusions who presented with less than 20% blasts and enlarged spleens and met the criteria for JMML.39 Optimal treatment regimens for these patients are not known, but we have elected to treat these patients with AML-like chemotherapy in combination with a targeted agent if applicable and proceed to stem cell transplantation after 1 to 2 cycles of therapy.
Secondary mutations outside the Ras pathway
Recurrent mutations found outside the canonical Ras pathway include alterations in epigenetic regulating genes, transcription factors, the spliceosome complex, and signal transduction pathways.13-15 Many PRC2 complex members have now been found to be mutated in JMML including ASXL1, DNMT3A, and EZH2. In a few cases, mutations in the spliceosome gene ZRSR2 have been identified. Also, transcription factors such as GATA2 and RUNX1 and signal transduction genes, including SH2B3 and JAK3, have been reported as recurrent mutations.13,14,40 Most importantly, mutations in SETBP1, whose function remains to be fully elucidated, are the most frequent secondary event and are associated with the worst prognosis, even when initially present as a subclonal event at diagnosis.40-42
Altered DNA methylation in JMML
Epigenetic alterations, particularly in DNA methylation, have been studied because of the lack of clinical or genetic markers that fully predict the diverse disease course of JMML and can serve as potential therapeutic targets. We and others demonstrated that altered methylation is a common feature in JMML, especially in patients with a more aggressive course of disease, and frequently accompanies the presence of secondary mutations.15,43,44 Whether alterations in DNA methylation allow for the acquisition of additional mutations or whether genetic mutations are causing an altered DNA methylation state is not completely understood. Methylation profiling of DNA extracted from newborn dried blood spots suggests that epigenetic changes are most likely a secondary event to genetic mutations.45 Clinical DNA methylation testing for patients with JMML will be available in the near future on multiple continents through an international collaboration, including our own group, wherein a consensus definition of methylation subgroups was developed based on data from more than 250 patients with JMML.46
Differential diagnoses mimicking JMML
NGS now allows for molecular confirmation of JMML in nearly all patients. However, in the absence of Ras pathway mutations, other diseases mimicking JMML need to be excluded. Myeloproliferative diseases including those with eosinophilia caused by constitutively activated platelet-derived growth factor α/β (PDGFR α/β) or fibroblast growth factor receptor 1 (FGFR1), are separate entities as defined by the WHO.47 Also, infants with KMT2A-rearranged acute leukemia can infrequently present with a low blast count and resemble JMML. Non-neoplastic disorders that are important to distinguish from JMML include viral infections, Wiskott-Aldrich syndrome, and infantile malignant osteopetrosis.48–50
Spectrum of malignancy in JMML
Ras-associated autoimmune leukoproliferative disorder (RALD) is a condition that overlaps with JMML and is similarly characterized by infants and toddlers who present with splenomegaly and somatic Ras mutations (although exclusively in KRAS and NRAS).51,52 In contrast to JMML, patients with RALD typically present with autoimmune sequelae and have an indolent clinical course related to their leukemia. Importantly, patients with RALD can still develop malignancies including progression to advanced JMML or lymphoma later in life.53 Considering that more than half of patients with JMML present with laboratory evidence of autoimmunity including Coombs antibodies and elevated levels of immunoglobulins, distinguishing JMML from RALD based on laboratory values alone can be challenging.6 Identifying patients with RALD using immunophenotypic evidence of circulating activated monocytes and polyclonal CD10+ B cells is not universally accepted.51 Further research delineating the differences between JMML and RALD is therefore warranted.
Risk stratification
Clinical and laboratory prognostic features
Unfavorable prognostic variables include older age at diagnosis (>2 years), platelet count < 33 × 109/L, and increased age-adjusted hemoglobin F levels. These factors consistently correlate with poor outcome (reduced event-free and overall survival) after HCT.54-56 Importantly, fetal hemoglobin is difficult to assess in infancy because of the wide range of normal values. Fetal hemoglobin is also rarely elevated in the presence of monosomy 7, which has been correlated with low levels of LIN28B expression and consequent high levels of BCL11A, a known transcription factor that suppresses γ globin production.57–59
Before NGS, it was recognized these clinical and laboratory features were associated with a worse prognosis; however, none of these factors independently cause a worse prognosis but rather are markers of those likely to have secondary mutations, an AML-type gene expression signature at diagnosis,60 or a hypermethylated DNA profile.
Prognostic implications of multiple genetic alterations
Several groups hypothesized that mutational status correlates with disease outcome. Although initial reports were conflicting, most groups have now found that the 5 canonical mutated genes, although uniformly initiating disease by Ras hyperactivation, are not independently prognostic of outcome.13,60 Instead, cooperating mutations are now recognized to contribute to progression of disease. These mutations include the aforementioned alterations in epigenetic regulating genes, transcription factors, the spliceosome complex, and signal transduction pathways. Patients with more than 1 mutation have a significantly worse prognosis, and recent reports suggest that these secondary mutations are frequently subclonal at diagnosis but emerge at the time of relapse.40,41 Detection of secondary events at diagnosis is thus critical to clinical management because a “watch-and-wait” approach is not appropriate in patients harboring additional mutations.
Using altered DNA methylation for risk stratification
Intimately tied to the presence of secondary mutations is the phenomenon of altered DNA methylation. Several groups have now shown that a hypermethylated DNA signature is present in patients with a higher risk of relapse, and these patients frequently have more than 1 genomic alteration. DNA hypermethylation has been independently associated with poor outcome in multivariable analysis even when age, fetal hemoglobin, and the number of mutations were included.43,46 In contrast, DNA hypomethylation is associated with a favorable diagnosis, and we described that patients with DNA methylation signatures similar to healthy age-matched subjects are those most likely to experience spontaneous resolution.15,43,44
In summary, more than traditional clinical or routine laboratory features, the presence of secondary mutations and/or a hypermethylated DNA signature are the factors most highly associated with an unfavorable prognosis. These patients should receive HCT and/or experimental clinical trials as the event-free survival for these patients is ~ 25% at 4 years.43 Conversely, rare patients with a single mutated gene (NRAS or KRAS) or patients with germline CBL syndrome who have a hypomethylated signature can likely be observed or treated with a hypomethylating agent without proceeding directly to HCT. These patients are typically infants who present with higher platelet counts and normal fetal hemoglobin.
Treatment
Indication for HCT vs watch-and-wait strategy
Our risk-stratified approach to treatment in the absence of a clinically available DNA methylation assay uses the number and type of mutations, age, and fetal hemoglobin and is summarized in Figure 1. Patients with germline CBL or PTPN11 mutations should be observed for spontaneous resolution. If failure to thrive is noted because of splenomegaly or the patient is symptomatic from leukocytosis, anemia, or thrombocytopenia, treatment with oral 6-mercaptopurine (50 mg/m2/d) with or without cis-retinoic acid (100 mg/m2/d) can alleviate symptoms, although it typically does not affect the molecular disease burden.61,62 The optimal duration of treatment of patients with CBL syndrome is still not known. Patients with a single mutation in NRAS or KRAS who are less than 1 year of age and have a normal fetal hemoglobin can likewise be initially treated with azacitidine (75 mg/m2/d) or be observed. It is not known whether observation or treatment with azacitidine in these patients is the optimal approach, and these patients fall into an intermediate category. In general, we favor treatment over observation if a patient is symptomatic because of cytopenias or splenomegaly. When treating with azacitidine, we typically continue therapy until a molecular remission is achieved, which can require multiple cycles. We recommend that these patients should only be transplanted if there is acquisition of additional mutations or clinical symptoms related to disease progression; therefore, close follow-up is critical. In our current treatment algorithm, patients with multiple mutations, age greater than 1 year, or elevated fetal hemoglobin receive moderate intensity therapy with fludarabine (30 mg/m2/d for 5 days) and cytarabine (2 g/m2/d for 5 days) and proceed to HCT after 1 to 2 cycles, ideally after molecular remission is achieved. Clinical response criteria to assess the efficacy of such therapies have also been defined.63 In the case that a remission is not achieved, patients may be considered for trials of experimental agents. Lacking those, patients should proceed to HCT, using all available strategies to augment the graft-versus-leukemia (GVL) effect.
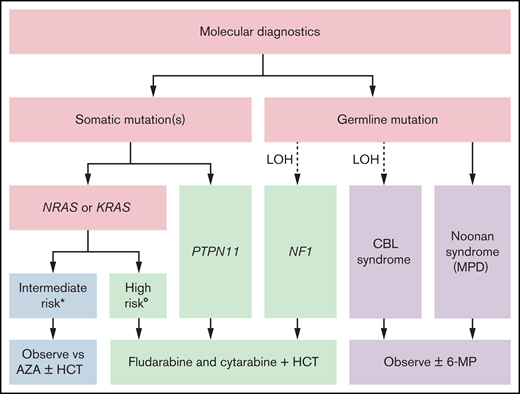
Risk stratified treatment algorithm as proposed by the authors. *All the following: single mutation, <1 year of age, and normal hemoglobin F. (Low DNA methylation when testing becomes clinically available.) °Any of the following: multiple mutations, >1 year of age, or elevated hemoglobin F. (Intermediate or high DNA methylation when testing becomes clinically available.) 6-MP, 6-mercaptopurine; AZA, azacitidine.
Risk stratified treatment algorithm as proposed by the authors. *All the following: single mutation, <1 year of age, and normal hemoglobin F. (Low DNA methylation when testing becomes clinically available.) °Any of the following: multiple mutations, >1 year of age, or elevated hemoglobin F. (Intermediate or high DNA methylation when testing becomes clinically available.) 6-MP, 6-mercaptopurine; AZA, azacitidine.
Pre-HCT and targeted therapy
Distinct from all other childhood leukemias, where a lower disease burden before HCT is associated with favorable outcomes, many patients with JMML have historically been taken to HCT with minimal or no pretransplant therapy.64 This practice was based on univariate analysis of data that suggested no difference in post-HCT survival in patients who did or did not receive moderately intense chemotherapy.65 However, this analysis may have been confounded by the inclusion of patients with >20% blasts (ie, AML arising from JMML). Subsequent analysis restricted to patients with typical JMML undergoing umbilical cord blood (UCB) transplant did demonstrate a survival benefit to pre-HCT moderate-intensity chemotherapy.66 We recently published our single institution retrospective study of 21 patients, of whom 40% received moderately intensive chemotherapy with intermediate-dose cytarabine and fludarabine and achieved a molecular response defined by a reduction of mutant allele frequency of the driver mutation to <5% before HCT. These patients had excellent outcomes after transplant.62 Most importantly, patients who entered transplant in molecular remission did not require extensive graft-versus-host disease (GVHD) to achieve a cure, whereas all patients who entered transplant with active disease relapsed in the absence of extensive GVHD. There are no randomized trials comparing pre-HCT therapy vs proceeding directly to HCT. Therefore, the optimal approach to treatment at diagnosis is not known.
Another approach to upfront treatment involves azacitidine. Intrigued by promising retrospective analyses,67 a recently completed clinical trial in Europe used azacitidine in 18 patients with newly diagnosed JMML (EudraCT #2014-002388-13). A preliminary analysis of this trial surprisingly revealed that patients most likely to respond had hypomethylated DNA signatures, suggesting that demethylation may not be the only mechanism of action of azacitidine.68 There are no randomized trials comparing different types of pre-HCT therapy.
There is also an ongoing trial sponsored by the Children’s Oncology Group for relapsed and refractory patients using the oral MEK inhibitor, trametinib, but data are not yet available for these patients (NCT03190915). It is reasonable to treat patients with fusions involving tyrosine kinases with relevant targeted treatments as part of their pre- and post-HCT course; however, because of the rarity of these events, supportive data are largely anecdotal.15,34,37
Finally, splenectomy before HCT was not found to have an impact on posttransplantation outcomes65 ; however, massive splenomegaly with hypersplenism and poor response to platelet transfusion are important clinical reasons to consider this procedure in selected cases, balancing immediate benefits with the long-term increased risk of overwhelming sepsis from encapsulated organisms in asplenic patients.
HCT
Most patients with JMML will ultimately require allogeneic HCT from the best available donor for cure of their disease. Beyond patients with spontaneous regression, the exception may be patients with CBL-mutated JMML. When following patients who have not been transplanted, these patients experience high rates of vascular complications that may be preventable by allogeneic HCT.17 The optimal approach for these patients remains unknown.
Because of the rarity of JMML, most data describe outcomes with matched sibling and adult unrelated donors.65 One report suggested poor outcomes with UCB donors66 compared with near-contemporary reports with traditional donors. The use of haploidentical donors for this condition is largely anecdotal.34,69 Therefore, UCB and haploidentical transplants should only be attempted at centers with expertise in those approaches. Although JMML-specific data are lacking, by analogy to AML, if multiple suitable donors are available, centers may consider killer immunoglobin-like receptors testing to potentially optimize natural killer cell–mediated GVL effects.70,71
Patients are frequently conditioned for HCT with an intensive triple-alkylator conditioning regimen, including busulfan, cyclophosphamide, and melphalan (Bu-Cy-Mel).61,65 This regimen is associated with high rates of sinusoidal obstruction syndrome. However, less intensive regimens need to be used with caution and perhaps should be reserved for those patients who achieve a molecular remission before transplant. The most recent Children’s Oncology Group trial (ASCT1221), which randomized patients to either Bu-Cy-Mel vs busulfan and fludarabine (Bu-Flu) closed early because of inferior outcomes in the Bu-Flu arm; however, it was noted retrospectively that there were more patients with adverse biologic risk factors enrolled on the Bu-Flu arm.61 A double-alkylator approach of busulfan, fludarabine, and melphalan has also been reported with good outcomes.72 Total body irradiation is currently avoided as part of first-line conditioning for patients because of the lack of apparent benefit over chemotherapy-based approaches and given the young age of these patients with the known late effects seen after total body irradiation.73,74
Given that most patients with JMML are only curable with allogeneic HCT, this suggests that disease control is primarily mediated by the alloreactive GVL effect. However, modulation of the GVL effect can be challenging, because it may be accompanied by potentially life-threatening GVHD. Therefore, the use of serotherapy, such as antithymocyte globulin, to prevent rejection and GVHD is controversial for patients with JMML, because there are conflicting reports in other malignant diseases about its possible role in increasing post-HCT relapse risk. We generally avoid the use of GM-CSF both before and after HCT, except in the case of life-threatening infections. This is not data driven but is based on the hypersensitivity of in vitro JMML cells to GM-CSF. Our HCT and post-HCT strategies are summarized in Figure 2.
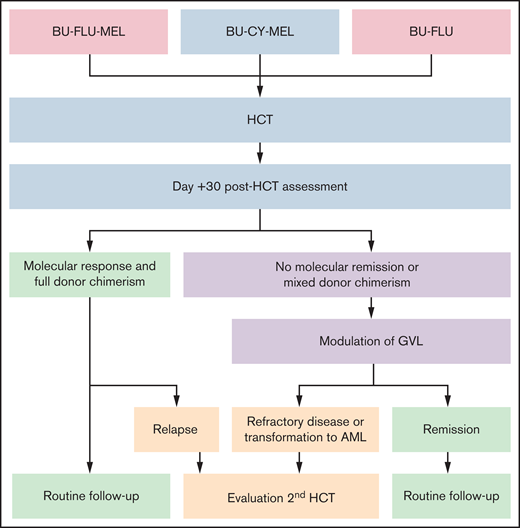
HCT and post-HCT strategy. Conditioning regiments include busulfan (BU; 16-20 mg/kg orally over 4 days) in combination with cyclophosphamide (CY; 120 mg/kg over 2 days) or fludarabine (FLU; 40 mg/m2/dose over 4 days) with or without melphalan (MEL; 140 mg/m2 once). Assessment at day +30 after HCT is performed by NGS and evaluation of donor chimerism. Molecular response is defined by a reduction of mutant allele frequency of the driver mutation to <5%. Modulation of GVL includes rapid withdrawal of immunosuppression and administration of donor lymphocyte infusions (±azacitidine). If remission, including molecular remission and full donor chimerism, is achieved, patients are monitored with bone marrow aspirates every 90 days for the first 18 months to 2 years after transplant (routine follow-up). Patients with refractory disease, relapse, or transformation to AML may benefit from a second HCT.
HCT and post-HCT strategy. Conditioning regiments include busulfan (BU; 16-20 mg/kg orally over 4 days) in combination with cyclophosphamide (CY; 120 mg/kg over 2 days) or fludarabine (FLU; 40 mg/m2/dose over 4 days) with or without melphalan (MEL; 140 mg/m2 once). Assessment at day +30 after HCT is performed by NGS and evaluation of donor chimerism. Molecular response is defined by a reduction of mutant allele frequency of the driver mutation to <5%. Modulation of GVL includes rapid withdrawal of immunosuppression and administration of donor lymphocyte infusions (±azacitidine). If remission, including molecular remission and full donor chimerism, is achieved, patients are monitored with bone marrow aspirates every 90 days for the first 18 months to 2 years after transplant (routine follow-up). Patients with refractory disease, relapse, or transformation to AML may benefit from a second HCT.
Post-HCT monitoring and therapy
Patients who are not in molecular remission before HCT are at very high risk for relapse. The modifying factor to this risk is the subsequent development of GVHD, which we and others have noted to be protective of relapse.62,72 We therefore assess all patients after engraftment, around day +30 after HCT, using NGS and sorted (myeloid and T-cell) donor chimerism. We anecdotally noted that occasional patients who have a positive, albeit low, percentage of their diagnostic mutation present at day +30 can achieve remission with the development of GVL (often accompanying clinical GVHD). Patients without GVHD who are not in molecular remission or who have mixed donor chimerism should undergo rapid withdrawal of immune suppression starting as early as day +30. If this is insufficient to achieve full donor chimerism in all cell compartments, including T cells, and thereby break donor tolerance to host hematopoietic elements, patients may benefit from receiving donor lymphocyte infusions with or without azacitidine.75–77
Most patients with JMML will ultimately require 100% donor chimerism in all lineages for cure, although patients with CBL mutations are possible exceptions.17,78 One theoretical benefit to entering HCT in molecular remission is that there is more time for the GVL effect to develop and provide ultimate disease control before residual disease inevitably progresses, as demonstrated in Figure 3.
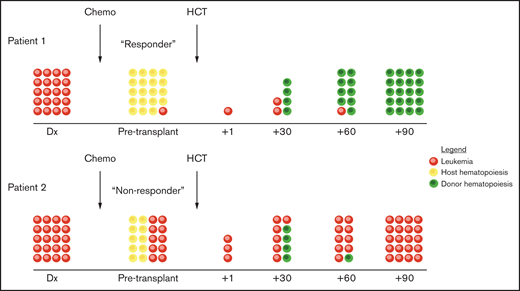
Patients can also potentially receive azacitidine after transplant along with donor lymphocyte infusions to try to enhance the GVL effect that is required to salvage patients who have molecular evidence of disease after transplant.79 This prophylactic approach has been shown to be potentially effective in adults with AML and myelodysplastic syndromes , but data in pediatrics and especially patients with JMML are lacking. We continue to monitor patients with bone marrow aspirate every 90 days for the first 18 months to 2 years after transplant, which is when most relapses occur. Patients who experience frank relapse can be salvaged with second transplants in ~ 30% to 40% of cases.65,80 Patients who transform to AML have dismal outcomes, with ~ 5% to 10% of patients experiencing long-term survival.60,65
Future directions
Molecular testing has dramatically enhanced our ability to diagnose patients with JMML. Mutation analysis at follow-up visits should be performed as standard of care to detect emerging cooperative events, especially in patients who do not proceed to HCT. Risk-stratified algorithms will improve with the introduction of clinically available DNA methylation testing and will allow for the identification of patients most likely to experience spontaneous resolution. Additional targeted agents directed toward the initiating Ras pathway mutations and secondary mutations in other genes including SETBP1 are still needed. Recent advances in patient-derived xenograft models and induced pluripotent stem cell models are new tools to identify and test the efficacy of these additional agents.81–83 Advances in chimeric antigen receptors may offer a paradigm shift in our approach to treatment but has yet to be trialed in JMML patients.84 Because of the rarity of this disease, international collaboration will be critical to test single agent and combinatorial regimens in our effort to move away from a reliance on intensive allogeneic HCT in these young patients.
Authorship
Contribution: A.W. and E.S. wrote the manuscript. C.C.D. and M.L.L. edited the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Elliot Stieglitz, University of California, San Francisco, CA; e-mail: stieglitze@peds.ucsf.edu.

