

Key Points
Clinically significant bleeding occurred in 9% of patients after CAR T therapy and was associated with features of systemic coagulopathy.
Low baseline platelets and possibly high-grade ICANS are risk factors for bleeding and require close monitoring for bleeding up to 1 month.

Abstract
Bleeding and thrombotic events are an emerging toxicity associated with chimeric antigen receptor (CAR) therapies. To determine their incidence, we retrospectively analyzed consecutive adult patients (N = 127) with large B-cell lymphoma (LBCL) or B-cell acute lymphoblastic leukemia (B-ALL) treated from 2017 through 2020 with axicabtagene ciloleucel (axi-cel; n = 89) or a bispecific CD19/CD22 CAR (n = 38). Twelve (9.4%) and 8 (6.3%) patients developed bleeding and thrombosis within the first 3 months, respectively. In the axi-cel subgroup, these occurred in 11.2% and 6.7%, respectively. Bleeding occurred between days 8 and 30 (median, 17.5) and thrombosis between days 2 and 91 (median, 29). Bleeding sites included genitourinary, soft tissue, intracranial, gastrointestinal, and pulmonary and were associated with features of consumptive coagulopathy. On univariate analysis, patients with bleeding were older, had lower baseline platelets (86 × 103/μL vs 178 × 103/μL; P < .01), lower platelet and fibrinogen nadirs , and elevated lactate dehydrogenase. Immune effector cell (IEC)–associated neurotoxicity syndrome (ICANS) grade ≥3 was associated with increased bleeding (50% vs 15%; P = .01), thrombosis (50% vs 16%; P = .04), prothrombin time prolongation, hypofibrinogenemia, and elevated D-dimer. Low pretreatment platelet counts were associated with bleeding in a multivariate logistic regression model. Patients with thrombocytopenia or severe ICANS are at increased risk of bleeding and should be closely monitored, particularly within the first month after CAR therapy. Future studies in larger cohorts should assess risk factors for systemic coagulopathies in CAR T therapy, including their association with neurotoxicity.
Introduction
Chimeric antigen receptor T-cell (CAR T) therapy has revolutionized the field of malignant hematology. CD19-targeted CAR T-cell therapy has resulted in effective, often durable, responses for chemotherapy-refractory B-cell lymphoma and B-acute lymphoblastic leukemia (B-ALL) in prospective clinical trials.1-5 CAR T cells against B-cell maturation antigen have also shown overall response rates in the range of 70% to 80% and very good partial response or better in 50% to 60% of heavily pretreated patients with multiple myeloma.6-8 As the CAR T experience matures, so does our recognition of emerging toxicities that may contribute to the morbidity of our patients. Among these, distinct hematologic disorders including impaired hematopoietic reconstitution,9,10 hemostatic defects,11,12 and a tendency toward thrombosis may be acquired after CAR T therapy.13 Whereas it is known that prolonged cytopenias can lead to prolonged transfusion dependency and infectious complications, less is understood about the consequences of abnormal hemostasis after CAR T therapy that could promote bleeding or thrombotic complications. Such coagulation abnormalities, specifically diffuse intravascular coagulation (DIC) are frequently attributed to cytokine release syndrome (CRS),11,14,15 although the mechanism and true relationship of such findings remains poorly understood. Ongoing exploration of the incidence and characteristics of bleeding and thrombosis after CAR T therapy is therefore necessary to elucidate such mechanisms and for the development of optimal management strategies.
In this retrospective study, we recorded bleeding episodes and thrombotic complications and their associated clinical and laboratory characteristics in a cohort of 127 consecutive patients with diffuse large B-cell lymphoma (DLBCL) or ALL who were treated with CD19-directed CAR T therapy at our institution.
Methods
Patient selection
The study adhered to our Institutional Review Board–approved database and biorepository protocol and was conducted in accordance with the Declaration of Helsinki. This is a retrospective, single-institution analysis of consecutive adult patients with relapsed or refractory DLBCL or B-ALL treated from 2017 through 2020 with CD19 CAR T therapy with either axicabtagene ciloleucel (axi-cel; n = 89) or a bispecific CD 19/22 CAR construct, using 4-1BB costimulatory domains (n = 38). All patients received lymphodepleting (LD) chemotherapy with cyclophosphamide (500 mg/m2) and fludarabine (30 mg/m2) on days −5 through −3, followed by CAR T-cell infusion on day 0. Patients received standard supportive and preventive measures including antiseizure prophylaxis with levetiracetam (days −1 through 28) and granulocyte colony-stimulating factor, starting on day +1 until a white blood cell count >5 × 103/μL is achieved and as needed afterward to maintain an absolute neutrophil count >1 × 103/μL. Platelet transfusions were given to maintain platelets ≤10 × 103/μL for asymptomatic patients, and ≤20 × 103/μL in the presence of any active bleeding. Higher transfusion thresholds were individualized at the discretion of physician providers based on severity of bleeding. Based on the evolving clinical experience with CAR T therapy, institutional guidelines recommend considering cryoprecipitate transfusion to maintain fibrinogen ≥100 μg/mL in asymptomatic patients.
Clinical data including laboratory measurements
Baseline characteristics and clinical events occurring after CAR T therapy were obtained by comprehensive review of patients’ electronic medical record data through the first 90 days after CAR T therapy. Platelet counts, coagulation parameters including thrombin time (TT), prothrombin time (PT), partial thromboplastin time (PTT), D-dimer, ferritin, CRP, and absolute monocyte counts were collected before lymphodepletion (pre-LD) on days 0, 3, 7, 14, 21, 28, 60, and 90. Medication administration records were reviewed to determine exposure to anticoagulant therapy, transfusions, and immunomodulatory therapy including glucocorticoids and anticytokine agents.
Adverse event and toxicity assessment
Bleeding events were included if graded ≥2 according to modified World Health Organization bleeding criteria, which have been commonly used in platelet transfusion trials.16-18 Thrombotic events were included in the analysis if graded ≥2 using Common Terminology Criteria for Adverse Events (v 5.0) or if a clinical intervention was required. CAR T toxicity assessment was monitored routinely through the first 30 days after infusion and continued as needed in patients who developed later complications. Cytokine release syndrome (CRS) and immune-effector cell (IEC)–associated neurotoxicity syndrome (ICANS) were graded by American Society for Transplantation and Cellular Therapy guidelines.19 Management of CRS and ICANS was carried out according to institutional guidelines.
Statistical analysis
Univariate analysis was implemented by Fisher’s exact test or χ2 test to describe associations between categorical variables and by the Mann-Whitney U test for continuous variables. Multivariable modeling was used to assess for associations between patient characteristics and bleeding events, and included 22 covariates that were selected via the GLMNET algorithm based on the LASSO regularization method of Friedman et al.20 The small number of bleeding and thrombotic events dictated that we apply Firth’s bias-corrected logistic regression model.21 When necessary, missing laboratory values were estimated by using regression imputation with available data. Because of the lower incidence of thrombosis, this model was not feasible for analysis of thrombotic events. Statistical analyses and plots were generated via GraphPad Prism (v 8), and R (v 3.6.1).
Results
Patient characteristics and the incidence of bleeding and thrombosis
One hundred twenty-seven patients with relapsed/refractory DLBCL (n = 111 [n = 89 received axi-cel and n = 22 received a bispecific CD19-CD22 CAR]) or B-ALL (n = 16 received a bispecific CD19-CD22 CAR) were included in the study. Median age was 61 years (range, 21-82) and median pre-LD platelet count was 174 × 103/μL (range, 8-483). Patients received a median of 3 prior lines of therapy (range, 2-12) including prior autologous or allogeneic transplant in 38 (30%) patients. Twenty-six (21%) patients also received bridging therapy before CAR T. Additional baseline patient characteristics are shown in Table 1, along with their association with bleeding and thrombosis events.
Baseline patient characteristics and their association with bleeding and thrombosis
| Patient baseline characteristics | Total | Bleeding | P | Thrombosis | P | ||
|---|---|---|---|---|---|---|---|
| Yes (n = 12) | No (n = 115) | Yes (n = 8) | No (n = 119) | ||||
| Sex, n (%) | .21 | .99 | |||||
| Male | 82 | 10 (83.3) | 72 (62.6) | — | 5 (62.5) | 77 (64.8) | — |
| Female | 45 | 2 (16.7) | 43 (37.4) | — | 3 (37.5) | 42 (35.2) | — |
| Age, y, median (range) | 61 (21-82) | 72 (58-77) | 60 (21-82) | <.01 | 67.5 (21-76) | 61 (25-82) | .47 |
| Diagnosis, n (%) | |||||||
| DLBCL | 111 | 11 (91.7) | 100 (87) | .99 | 7 (87.5) | 104 (87.4) | .99 |
| ALL | 16 | 1 (8.3) | 15 (13) | — | 1 (12.5) | 15 (12.6) | — |
| Disease burden markers, n (%) | |||||||
| Elevated baseline LDH | 61 | 10 (83.3) | 51 (44.3) | .01 | 4 (50) | 57 (47.9) | .99 |
| BM blasts (ALL) >5%, n (%) | 7 | 1 (8.3) | 6 (5.2) | .51 | 1 (12.5) | 6 (5) | .37 |
| Baseline platelets, ×103/μL, median (range) | 174 (8-483) | 86 (8-219) | 178 (24-483) | <.01 | 207 (134-311) | 174 (8-483) | .11 |
| Prior lines of therapy, n median (range) | 3 (2-12) | 3 (2-4) | 3 (2-12) | .71 | 3 (2-5) | 3 (2-12) | .29 |
| Prior transplant, n (%)* | 38 | 2 (16.7) | 36 (31.3) | .51 | 1 (12.5) | 37 (31.1) | .43 |
| Bridging therapy, n (%) | 26 | 5 (41.7) | 21 (18.3) | .07 | 3 (37.5) | 23 (19.3) | .36 |
| History of bleeding, n | 4 | 3 (25) | 1 (0.8) | <.01 | 0 (0) | 4 (3.4) | .99 |
| History of clotting, n | 25 | 4 (33.3) | 21 (18.3) | .25 | 3 (37.5) | 22 (18.4) | .19 |
| Neurologic comorbidity, n | 7 | 1 (8.3) | 6 (5.2) | .51 | 2 (25) | 5 (4.2) | .06 |
| Vascular comorbidity, n | 12 | 2 (16.7) | 10 (8.7) | .32 | 2 (25) | 10 (8.4) | .17 |
| Anticoagulation (preceding CAR T), n | 9 | 1 (8.3) | 8 (7) | .99 | 0 (0) | 9 (7.6) | .99 |
| Patient baseline characteristics | Total | Bleeding | P | Thrombosis | P | ||
|---|---|---|---|---|---|---|---|
| Yes (n = 12) | No (n = 115) | Yes (n = 8) | No (n = 119) | ||||
| Sex, n (%) | .21 | .99 | |||||
| Male | 82 | 10 (83.3) | 72 (62.6) | — | 5 (62.5) | 77 (64.8) | — |
| Female | 45 | 2 (16.7) | 43 (37.4) | — | 3 (37.5) | 42 (35.2) | — |
| Age, y, median (range) | 61 (21-82) | 72 (58-77) | 60 (21-82) | <.01 | 67.5 (21-76) | 61 (25-82) | .47 |
| Diagnosis, n (%) | |||||||
| DLBCL | 111 | 11 (91.7) | 100 (87) | .99 | 7 (87.5) | 104 (87.4) | .99 |
| ALL | 16 | 1 (8.3) | 15 (13) | — | 1 (12.5) | 15 (12.6) | — |
| Disease burden markers, n (%) | |||||||
| Elevated baseline LDH | 61 | 10 (83.3) | 51 (44.3) | .01 | 4 (50) | 57 (47.9) | .99 |
| BM blasts (ALL) >5%, n (%) | 7 | 1 (8.3) | 6 (5.2) | .51 | 1 (12.5) | 6 (5) | .37 |
| Baseline platelets, ×103/μL, median (range) | 174 (8-483) | 86 (8-219) | 178 (24-483) | <.01 | 207 (134-311) | 174 (8-483) | .11 |
| Prior lines of therapy, n median (range) | 3 (2-12) | 3 (2-4) | 3 (2-12) | .71 | 3 (2-5) | 3 (2-12) | .29 |
| Prior transplant, n (%)* | 38 | 2 (16.7) | 36 (31.3) | .51 | 1 (12.5) | 37 (31.1) | .43 |
| Bridging therapy, n (%) | 26 | 5 (41.7) | 21 (18.3) | .07 | 3 (37.5) | 23 (19.3) | .36 |
| History of bleeding, n | 4 | 3 (25) | 1 (0.8) | <.01 | 0 (0) | 4 (3.4) | .99 |
| History of clotting, n | 25 | 4 (33.3) | 21 (18.3) | .25 | 3 (37.5) | 22 (18.4) | .19 |
| Neurologic comorbidity, n | 7 | 1 (8.3) | 6 (5.2) | .51 | 2 (25) | 5 (4.2) | .06 |
| Vascular comorbidity, n | 12 | 2 (16.7) | 10 (8.7) | .32 | 2 (25) | 10 (8.4) | .17 |
| Anticoagulation (preceding CAR T), n | 9 | 1 (8.3) | 8 (7) | .99 | 0 (0) | 9 (7.6) | .99 |
N = 127. Categorical variables were analyzed by Fisher’s exact test and continuous variables by the Mann-Whitney U test. Bold indicates statistically significant results. P values in bold are .05 or less.
BM, bone marrow; NHL, non-Hodgkin lymphoma.
Autograft (n = 26); allograft (n = 12).
Twelve (9.4%) and 8 (6.3%) patients developed study-defined bleeding and thrombotic complications in the first 3 months after CAR T infusion, respectively (Figure 1A). Grade 1 events were excluded unless an intervention was necessary. These included self-limiting hemorrhoidal bleeding (n = 3), epistaxis (n = 1), and oral bleeding (n = 2). One patient with self-limiting superficial venous thrombosis was also excluded. All study-defined events are characterized in Figure 1B. All bleeding events occurred within the first month (median day, 17.5; range, 8-30) after CAR T therapy, and 7 (58%) occurred after discharge from the hospital. Events included gross hematuria (n = 6), soft tissue bleeding (n = 2), subdural hematoma (n = 2), gastrointestinal hemorrhage (n = 1), and hemoptysis (n = 1). Two patients with gross hematuria had indwelling catheters placed for urinary retention >5 days before the bleeding episode, although trauma was not thought to be a primary source of active hemorrhage based on clinical judgement. Two patients also had BK virus detectable in the urine before the bleeding event. Thrombotic events were observed for up to 3 months (median, day 29; range, 2-91) after CAR T therapy and included deep venous thrombosis (n = 5), thrombotic stroke (n = 1), and splanchnic vein thrombosis (n = 2). Two (25%) of these events were associated with central venous catheters, and 3 (38%) were found concurrently with progressive disease. Two (1.6%) patients experienced both bleeding and thrombosis. Of these, 1 experienced bleeding and thrombosis events concurrently on postinfusion day 8, and 1 experienced the events separately, on days 24 and 46, respectively. Older age (median, 72 vs 60 years; P < .01), elevated pre-LD lactate dehydrogenase (LDH; 83% vs 44%, P = .01) and a lower baseline platelet count (median, 86 × 103/μL vs 178 × 103/μL; P < .01) were all associated with a higher frequency of bleeding events on univariate analysis. A documented history of bleeding was also more frequent in patients with post–CAR T bleeding events than in those without bleeding (25% vs 1%; P < .01). Twenty-five patients had a documented history of thrombosis before CAR T infusion. Thrombotic events after CAR T therapy were seen in 3 (12%) patients with a history of thrombosis, compared with 5 (5%) patients without a history of thrombosis. This difference was not statistically significant in our cohort (P = .19), but this comparison is limited by the small sample size. The only baseline characteristic that maintained significance related to bleeding events in the multivariate model was thrombocytopenia before starting LD (P = .02; supplemental Table 1).
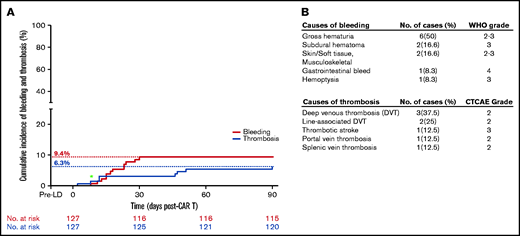
Incidence of bleeding and thrombotic events after CAR T therapy. (A) Cumulative incidence of bleeding and thrombosis. Green asterisk: a patient who had both bleeding and clotting events on the same day. (B) Tables characterizing the bleeding and thrombosis events and their respective clinical grades according to World Health Organization (bleeding) and Common Terminology Criteria for Adverse Events criteria (thrombosis). Events were included if the grade was >2 or if a clinical intervention was necessary.
Incidence of bleeding and thrombotic events after CAR T therapy. (A) Cumulative incidence of bleeding and thrombosis. Green asterisk: a patient who had both bleeding and clotting events on the same day. (B) Tables characterizing the bleeding and thrombosis events and their respective clinical grades according to World Health Organization (bleeding) and Common Terminology Criteria for Adverse Events criteria (thrombosis). Events were included if the grade was >2 or if a clinical intervention was necessary.
Anticoagulation did not increase the risk of bleeding after CAR T therapy
Thirteen patients (10% of study cohort) received anticoagulation for prophylactic or therapeutic indications that predated CAR T infusion. Four additional patients started anticoagulation in response to thrombotic events after CAR T infusion (n = 2; <30 days after infusion; n = 2; >30 days after infusion), and 1 received tissue plasminogen activator for an acute stroke. Among patients who received early anticoagulation after CAR T infusion (<30 days), 4 (27%) required temporary cessation due to thrombocytopenia, and 11 (73%) were continued safely through day 30. In our cohort, none of the bleeding events occurred in patients receiving anticoagulation therapy, and no patients developed bleeding complications after starting anticoagulation. In addition, no patients developed a thrombotic event after stopping anticoagulation.
Laboratory abnormalities associated with bleeding and thrombosis
Bleeding events.
Bleeding events (days 0-30) coincided with the onset of thrombocytopenia and hypofibrinogenemia in our cohort. Patients with bleeding had a lower platelet nadir after CAR T (median, 17.5 × 103/μL vs 48 × 103/μL; P < .01) and a lower fibrinogen nadir (median, 122 μg/mL vs 340 μg/mL; P < .01), compared with those without bleeding. Temporally, the lowest median platelet nadir occurred at day 7 in patients with bleeding events vs day 21 in patients without bleeding, whereas the median onset of fibrinogen nadirs was at day 21 in both groups. Other abnormal laboratory results in the first 30 days, seen more frequently in patients with bleeding, included PT prolongation >3 seconds (36% vs 8%; P = .02) and TT prolongation (trend, 70% vs 40%; P = .10). PTT prolongation >5 seconds (20% vs 27%; P > .99) and D-dimer elevation >3 times the upper limit of normal (ULN) (70% vs 73%; P > .99) were similar in patients with bleeding, compared with those without bleeding (Figure 2; Table 2). According to International Society on Thrombosis and Haemostasis (ISTH) criteria for DIC,22 patients with bleeding events did not have an increased incidence of overt DIC during the first 30 days, compared with patients without bleeding episodes (50% vs 35%; P = .48). Patients with bleeding events had a higher peak ferritin (median, 2523 ng/mL vs 1307 ng/mL; P = .01) and a lower absolute monocyte count (median, 0.48 × 103/μL vs 0.75 × 103/μL; P = .05), compared with those without bleeding events. Peak CRP (12.7 mg/dL vs 7.6 mg/dL; P = .15) was not significantly different in patients who had bleeding episodes compared with those who did not. One patient with a bleeding event had an elevation in total bilirubin ≥2 times the ULN. No patients with bleeding events had elevation in liver transaminases ≥3 times ULN, and in the majority (58%), the transaminases remained within normal limits. No patients had recent exposure to agents known to cause direct liver toxicity or hypofibrinogenemia including fibrinolytic agents. Ten patients (9%) without bleeding events in our study cohort received cryoprecipitate infusions according to protocols defined in “Methods.” Individual plots of platelet and fibrinogen trends and transfusion requirements in patients with bleeding events are shown in Figure 3.

Laboratory findings in patients with bleeding, thrombosis, and ICANS. Median platelet counts, fibrinogen, absolute monocytes, and ferritin in patients with bleeding events, thrombotic events, and ICANS during the first 90 days after CAR T infusion. Shaded areas represent pre-LD measurements (brown) and normal (gray) or abnormal (pink) ranges of laboratory values. Error bars, 95% CI.
Laboratory findings in patients with bleeding, thrombosis, and ICANS. Median platelet counts, fibrinogen, absolute monocytes, and ferritin in patients with bleeding events, thrombotic events, and ICANS during the first 90 days after CAR T infusion. Shaded areas represent pre-LD measurements (brown) and normal (gray) or abnormal (pink) ranges of laboratory values. Error bars, 95% CI.
Laboratory abnormalities in patients with bleeding and ICANS
| Laboratory findings | Total | Bleeding | P | ICANS Max Grade | P | ||
|---|---|---|---|---|---|---|---|
| Evaluable | Yes (n = 12) | No (n = 115) | 0-2 (n = 104) | 3-4 (n = 23) | |||
| Coagulation parameters | |||||||
| Prothrombin time (PT), n (%) | 96 (75.6) | — | — | .02 | — | — | .01 |
| 0-3-s prolongation | — | 7 (63.6) | 78 (91.8) | 70 (93.3) | 15 (71.4) | — | |
| >3-s prolongation | — | 4 (36.4) | 7 (8.2) | 5 (6.7) | 6 (28.6) | — | |
| PTT prolongation, n (%) | 74 (58.3) | — | — | >.99 | — | — | .36 |
| 0-5 s prolongation | — | 8 (80) | 47 (73.4) | — | 39 (70.9) | 16 (84.2) | — |
| >5 s prolongation | — | 2 (20) | 17 (26.6) | — | 16 (29.1) | 3 (15.8) | — |
| TT prolongation, n (%) | 58 (45.7) | 7 (70) | 19 (39.6) | .10 | 14 (31.8) | 12 (85.7) | <.01 |
| D-dimer elevation, n (%) | 61 (48) | — | — | >.99 | — | — | .19 |
| 0-3 x ULN | — | 3 (30) | 14 (27.5) | — | 15 (33.3) | 2 (12.5) | — |
| >3 x ULN | — | 7 (70) | 37 (72.5) | — | 30 (66.7) | 14 (87.5) | — |
| D-dimer peak, μg/mL, median (range) | 2.56 (0.77-20) | 3.76 (0.82-20) | 2.32 (0.77-20) | .48 | 2.22 (0.77-20) | 4.28 (0.87-20) | .02 |
| Platelet nadir, ×103/μL, median (range) | 40 (5-158) | 17.5 (6-52) | 48 (5-158) | <.01 | 53.5 (5-158) | 17 (9-75) | <.01 |
| Fibrinogen nadir, mg/dL, median (range) | 307.5 (66-1057) | 122 (66-589) | 340 (78-1057) | <.01 | 346 (78-1057) | 114.5 (66-939) | <.01 |
| ISTH DIC score ≥5, n (%) | 61 (48) | 5 (50) | 18 (35.3) | .48 | 14 (31.1) | 9 (56.3) | .13 |
| Inflammatory markers | |||||||
| Ferritin peak, ng/mL, median (range) | 1393 (84-65 881) | 2523 (652-65 881) | 1307 (84-62 092) | .01 | 1314 (84-62 092) | 1667 (560-65 881) | .07 |
| CRP peak, mg/dL, median (range) | 7.6 (0.4-60) | 12.7 (4-38) | 7.6 (0.4-60) | .15 | 7.4 (0.4-60) | 13.3 (2.4-60) | .01 |
| Laboratory findings | Total | Bleeding | P | ICANS Max Grade | P | ||
|---|---|---|---|---|---|---|---|
| Evaluable | Yes (n = 12) | No (n = 115) | 0-2 (n = 104) | 3-4 (n = 23) | |||
| Coagulation parameters | |||||||
| Prothrombin time (PT), n (%) | 96 (75.6) | — | — | .02 | — | — | .01 |
| 0-3-s prolongation | — | 7 (63.6) | 78 (91.8) | 70 (93.3) | 15 (71.4) | — | |
| >3-s prolongation | — | 4 (36.4) | 7 (8.2) | 5 (6.7) | 6 (28.6) | — | |
| PTT prolongation, n (%) | 74 (58.3) | — | — | >.99 | — | — | .36 |
| 0-5 s prolongation | — | 8 (80) | 47 (73.4) | — | 39 (70.9) | 16 (84.2) | — |
| >5 s prolongation | — | 2 (20) | 17 (26.6) | — | 16 (29.1) | 3 (15.8) | — |
| TT prolongation, n (%) | 58 (45.7) | 7 (70) | 19 (39.6) | .10 | 14 (31.8) | 12 (85.7) | <.01 |
| D-dimer elevation, n (%) | 61 (48) | — | — | >.99 | — | — | .19 |
| 0-3 x ULN | — | 3 (30) | 14 (27.5) | — | 15 (33.3) | 2 (12.5) | — |
| >3 x ULN | — | 7 (70) | 37 (72.5) | — | 30 (66.7) | 14 (87.5) | — |
| D-dimer peak, μg/mL, median (range) | 2.56 (0.77-20) | 3.76 (0.82-20) | 2.32 (0.77-20) | .48 | 2.22 (0.77-20) | 4.28 (0.87-20) | .02 |
| Platelet nadir, ×103/μL, median (range) | 40 (5-158) | 17.5 (6-52) | 48 (5-158) | <.01 | 53.5 (5-158) | 17 (9-75) | <.01 |
| Fibrinogen nadir, mg/dL, median (range) | 307.5 (66-1057) | 122 (66-589) | 340 (78-1057) | <.01 | 346 (78-1057) | 114.5 (66-939) | <.01 |
| ISTH DIC score ≥5, n (%) | 61 (48) | 5 (50) | 18 (35.3) | .48 | 14 (31.1) | 9 (56.3) | .13 |
| Inflammatory markers | |||||||
| Ferritin peak, ng/mL, median (range) | 1393 (84-65 881) | 2523 (652-65 881) | 1307 (84-62 092) | .01 | 1314 (84-62 092) | 1667 (560-65 881) | .07 |
| CRP peak, mg/dL, median (range) | 7.6 (0.4-60) | 12.7 (4-38) | 7.6 (0.4-60) | .15 | 7.4 (0.4-60) | 13.3 (2.4-60) | .01 |
Frequency of treatment-emergent abnormal laboratory values and median nadir or peak values in patients with bleeding, and ICANS grades 0-2 vs grades 3-4. Total evaluable represents the total number (for categorical variables) or median value (for continuous variables) from patients with available data for analysis. Patients without data available were excluded from the analysis. Normal range for D-dimer was <0.50 μg/mL fibrinogen-equivalent units. ISTH DIC score includes platelets (>100 = 0, 50-100 = 1, and <50 = 2), PT prolongation (< 3 s = 0< 3 s to <6 s = 1, ≥6 s = 2), D-dimer (0, no increase, <5 times ULN = 2, ≥5 times ULN = 3), and fibrinogen (>100 μg/mL = 0, ≤100 μg/mL=1) and a score ≥5 is suggestive of overt DIC. P for categorical variables was calculated by Fisher’s exact test and for continuous variables by the Mann-Whitney U test. P values in bold are .05 or less.

Individual bleeding events after CAR T infusion. Individual plots for each patient with bleeding events illustrating platelet and fibrinogen measurements, as well as platelet and plasma transfusion requirements during the first 30 days after CAR T infusion. Duration and maximum grade of CRS and ICANS are included. Patients are shown in sequential order based on timing of the bleeding event. Note that plots in patients without available fibrinogen measurements (n = 2) do not include gray lines. *Patients treated with a bispecific CD 19/22 CAR construct (n = 2), whereas the remaining patients (n = 10) received CD 19 CAR T therapy with axi-cel. GIB, gastrointestinal bleeding; GUB, genitourinary bleeding; SDH, subdural hematoma.
Individual bleeding events after CAR T infusion. Individual plots for each patient with bleeding events illustrating platelet and fibrinogen measurements, as well as platelet and plasma transfusion requirements during the first 30 days after CAR T infusion. Duration and maximum grade of CRS and ICANS are included. Patients are shown in sequential order based on timing of the bleeding event. Note that plots in patients without available fibrinogen measurements (n = 2) do not include gray lines. *Patients treated with a bispecific CD 19/22 CAR construct (n = 2), whereas the remaining patients (n = 10) received CD 19 CAR T therapy with axi-cel. GIB, gastrointestinal bleeding; GUB, genitourinary bleeding; SDH, subdural hematoma.
Thrombosis events.
Patients with thrombotic events were not observed to have significant differences in coagulation parameter abnormalities compared with patients without thrombosis, including the rate of PT prolongation >3 seconds (29% vs 10%; P = .18), PTT prolongation >5 seconds (0% vs 28%; P = .18), TT prolongation (67% vs 46%; P = .59), D-dimer elevation >3 times ULN (80% vs 71%; P > .99), or peak D-dimer (median, 4.97 μg/mL vs 2.46, P = .22). Patients with thrombotic events did show trends toward a higher peak CRP (15.3 mg/dL vs 7.6 mg/dL; P = .07) and lower peak absolute monocyte counts (0.61 × 103/μL vs 0.75 × 103/μL; P = .08).
Association of bleeding and thrombotic events with CRS and ICANS
Bleeding and thrombotic events were not associated with the occurrence or severity of CRS, although only 3 patients developed grade 3 or higher CRS in our cohort. Likewise, CRS was not associated with marked coagulation abnormalities. However, high-grade ICANS (grade ≥3) vs grades 0 to 2 ICANS was associated with incidences of bleeding (50% vs 15%; P = .01) and thrombosis (50% vs 16%; P = .04; Table 3). We observed a higher frequency of PT prolongation >3 seconds (29% vs 7%; P = .01), TT prolongation (86% vs 32%; P < .01), peak D-dimer (median, 4.28 μg/mL vs 2.22 μg/mL; P = .02), lower platelet count (17 × 103/μL vs 53.5 × 103/μL; P < .01), and fibrinogen (114.5 mg/dL vs 346 mg/dL; P < .01) nadirs in patients with high-grade ICANS. Although we observed a trend toward a higher incidence of overt DIC, according to ISTH DIC scoring criteria in patients with high-grade ICANS, compared with those with low-grade or no ICANS (56% vs 31%, P = .13); the increase was not statistically significant (Table 2). Patients with high-grade ICANS, similar to those with bleeding events, also showed more dramatic depletion of fibrinogen measured by the percentage change between pre-LD or day 0 fibrinogen and subsequent nadirs in patients who had available values across this time period (supplemental Figure 2).
Association of CRS and ICANS with incidence of adverse bleeding and thrombosis events
| Total (N = 127) | Bleeding | P | Thrombosis | P | |||
|---|---|---|---|---|---|---|---|
| Yes (n = 12) | No (n = 115) | Yes (n = 8) | No (n = 119) | ||||
| CRS maximum grade n, (%) | .26 | .18 | |||||
| Grade 0-2 | 124 (97.6) | 11 (91.7) | 113 (98.3) | — | 7 (87.5) | 117 (98.3) | — |
| Grade 3-4 | 3 (2.4) | 1 (8.3) | 2 (1.7) | — | 1 (12.5) | 2 (1.7) | — |
| ICANS maximum grade n, (%) | .01 | .04 | |||||
| Grade 0-2 | 104 (81.9) | 6 (50) | 98 (85.2) | — | 4 (50) | 100 (84) | — |
| Grade 3-4 | 23 (18.1) | 6 (50) | 17 (14.8) | — | 4 (50) | 19 (16) | — |
| Total (N = 127) | Bleeding | P | Thrombosis | P | |||
|---|---|---|---|---|---|---|---|
| Yes (n = 12) | No (n = 115) | Yes (n = 8) | No (n = 119) | ||||
| CRS maximum grade n, (%) | .26 | .18 | |||||
| Grade 0-2 | 124 (97.6) | 11 (91.7) | 113 (98.3) | — | 7 (87.5) | 117 (98.3) | — |
| Grade 3-4 | 3 (2.4) | 1 (8.3) | 2 (1.7) | — | 1 (12.5) | 2 (1.7) | — |
| ICANS maximum grade n, (%) | .01 | .04 | |||||
| Grade 0-2 | 104 (81.9) | 6 (50) | 98 (85.2) | — | 4 (50) | 100 (84) | — |
| Grade 3-4 | 23 (18.1) | 6 (50) | 17 (14.8) | — | 4 (50) | 19 (16) | — |
In addition, we observed a lower median absolute monocyte peak (median, 0.58 × 103/μL vs 0.75 × 103/μL; P = .01), higher CRP peak (median, 13.3 mg/dL vs 7.4 mg/dL; P = .01) and a trend toward higher ferritin peak (median, 1667 ng/mL vs 1314 ng/mL; P = .07) in patients with high-grade ICANS, compared with those with grades 0 to 2 (Table 2).
Multivariate analysis
We attempted to estimate the probability of bleeding events occurring based on a multivariable logistic regression model that included the following 22 baseline covariates: sex, disease, age, prior lines of therapy, prior transplant, bridging therapy, bleeding history, thrombosis history, history of vascular disease, neurologic comorbidities, anticoagulation before CAR T therapy, maximum grade CRS and ICANS, baseline monocytes, platelets, LDH, and coagulation abnormalities including PTT, PT, fibrinogen, TT, and D-dimer. Multivariate analysis using the GLMNET algorithm and Firth’s bias-corrected logistic regression model described in “Methods” were chosen, as they fit a model with a small number of events.20 We rescaled the continuous variable platelets by 10 to render the estimated model coefficient to be consistent with clinical experience. When we applied the variable selection and fit the model for the bleeding events, we found that baseline thrombocytopenia was statistically significant (P = .0024; supplemental Table 1). Because of the lower number of thrombotic events, logistic regression using Firth’s bias correction did not fit the model to the thrombotic events data.
Subgroup analysis of patients treated with axi-cel
To account for different CAR constructs in our study, we performed a subgroup analysis of patients treated with axi-cel (n = 89) and observed a higher incidence of bleeding (11.2%) and thrombosis (6.7%) events (supplemental Figure 3). Univariate characteristics and laboratory findings associated with these events were reflective of the entire study cohort (supplemental Figure 4; supplemental Tables 2 and 3) with the exception that differences in fibrinogen nadir (median,142.5 mg/dL vs 270 mg/dL; P = .12) in patients with bleeding events were not statistically significant. Whether this finding represents unique endovascular processes that occur after axi-cel is difficult to conclude in analyzing a small number of events. Similar to our entire study cohort, in multivariate analysis of characteristics in the axi-cel subgroup, baseline thrombocytopenia was the only significant predictor of subsequent bleeding events (supplemental Table 4B).
Discussion
We observed that both bleeding and thrombotic complications can occur in the first 3 months after CAR T therapy, with bleeding events being more common in the first month after CAR T therapy and patients with a low baseline platelet count and high-grade ICANS being at highest risk for bleeding events after CAR T therapy. This finding was supported in results of all patients undergoing CD19 directed CAR T therapy, including axi-cel and our institutional CD19-22 CAR T construct. Similar findings were observed in the subgroup of patients with lymphoma who were receiving axi-cel.
Although fibrinogen depletion and prolonged thrombocytopenia are commonly observed after CAR T therapy,9,11 their clinical sequelae have so far remained relatively unknown, particularly the potential for bleeding, thromboembolism, and other consequences of vascular compromise. Consistent with our report, recent findings suggest that patients with LBCL have an increased incidence of venous thromboembolism after treatment with CD19 CAR T therapy. However, increased bleeding complications were not observed in this study.13 In our cohort, we observed both bleeding (9.4%) and thrombotic (6.3%) events in the first 3 months after CAR T-cell therapy. All bleeding events occurred within the first 30 days after CAR T infusion, and more than 50% occurred after patients were discharged, illustrating the importance of close follow-up throughout this period to identify bleeding complications and to provide supportive measures including blood product transfusions when necessary. All events in our study were deemed clinically significant based on a requisite for clinical intervention or transfusion dependence related to active hemorrhage. We attempted to evaluate factors that increase the risk of bleeding. Although several baseline characteristics were associated with bleeding risk on univariate analysis (age, low baseline platelet count, and high tumor burden, as indicated by elevated LDH and history of bleeding), low baseline platelet count was the only factor predictive of increased bleeding risk on multivariate analysis. Our ability to find additional predictive risk factors in a multivariate model was limited by the number of bleeding events (eg, only 12 events) within our cohort. Treatment-emergent characteristics that were associated with increased risk of bleeding included grade 3 or higher ICANS, although this did not retain significance in a multivariate model. CRS was not associated with increased risk of bleeding; however, only 3 patients had high-grade CRS. Our findings suggest that patients with low baseline platelets and perhaps a history of grade 3 or higher ICANS should be particularly closely monitored with complete blood count and fibrinogen levels, even after hospital discharge until 1 month after CAR T therapy, as these patients have the highest risk of bleeding. Elective procedures or interventions including the use of foley catheters should also be minimized. The involved sites of bleeding observed after CAR T therapy included both mucocutaneous and deep tissues, reflecting a typical pattern of both primary and secondary hemostatic defects. This observation suggests that thrombocytopenia alone is unlikely to be sufficient to provoke these events, and a secondary event that triggered consumptive coagulopathy most likely added to increased risk in these patients. In our study, patients with bleeding had lower fibrinogen nadirs and more frequent PT prolongation. However, significant differences in D-dimer elevation were not seen, and although application of ISTH DIC criteria showed a high overall incidence of overt DIC, it was not associated with bleeding in our cohort, making it difficult to draw inferences about the bleeding risk associated with DIC after CAR T therapy. It is likely that DIC after CAR T therapy is akin to tumor lysis syndrome in patients with cancer,23 which can be divided into chemical and clinical tumor lysis syndromes, with the former being more common and only having laboratory abnormalities and the latter being associated with clinical manifestations and organ damage. It is likely that most cases of DIC with CAR T therapy are mild and not associated with clinical bleeding. Moreover, our institutional policy is to consider prophylactic cryoprecipitate administration in asymptomatic patients when fibrinogen is <100 mg/dL. This prophylactic strategy possibly prevents clinically significant bleeding in some patients, although our data do not provide definite evidence to support that.
Although bleeding events have not been reported in the pivotal CD19 CAR T trials for adults with LBCL and ALL,1,3,5 1 fatality caused by cerebral hemorrhage was reported after tisagenlecleucel in pediatric B-ALL,4 and 4 bleeding events graded 3 to 4 were observed in a recent phase 2 study of idecabtagene vicleucel, a B-cell maturation antigen–directed CAR T trial for patients with refractory myeloma.8 Clinically significant bleeding after other intense hematologic therapies, such as autologous and allogeneic hematopoietic cell transplantation are reported to occur in between 2% and 80% of cases.24-29 Measures of DIC incidence in these settings are less precise, but may generally occur in 10% to 20% of myeloid (excluding Acute promyelocytic leukemia) or lymphoid malignancies with a preponderance of thrombotic rather than bleeding complications.30-32
Our findings suggest that a systemic coagulopathy occurs after CAR T therapy, coinciding with predominant bleeding manifestations. However, despite a high incidence of DIC according to standard criteria this itself does not appear to modulate the risk of bleeding in our study. This finding raises questions about the generalizability of routinely used scoring algorithms for DIC in CAR T settings and may imply that biologic processes mediating CAR T–related coagulopathy are more distinct from other known disorders associated with DIC. As described herein, it is also possible that the DIC-like picture is mild in most cases and not associated with clinical sequelae such as bleeding. We also hypothesize that the biphasic thrombocytopenia observed in our cohort involves at least 2 distinct processes: the first phase being attributable to LD chemotherapy, and the second being less understood, but most likely because of CAR T–related bone marrow suppression.33 Importantly, both mechanisms of thrombocytopenia compete with true platelet consumption in the interpretation of DIC scoring algorithms, minimizing their specificity for DIC after CAR T therapy. Likewise, D-dimer elevation after CAR T therapy may be confounded by multiple factors including active malignancy, presence of central venous catheters, venous thromboembolic disease, systemic inflammation, and acute bleeding. We therefore propose that standard scoring algorithms for DIC may be inadequate to portray distinct CAR T–related coagulopathy and its clinical manifestations, and the development of more specific criteria may promote a better understanding of CAR T–related coagulopathy.
In our cohort, patients were supported with blood products including platelet and/or cryoprecipitate and fresh-frozen plasma infusions, which was sufficient to resolve their active hemorrhage. However, whether the intravascular biology that triggers bleeding in such patients is truly reminiscent of classic DIC remains unknown, as does the optimal management strategy for active bleeding after CAR T therapy. It is plausible that direct activity of CAR T cells or their cytokine mediators promotes coagulation factor consumption, endothelial activation, and propagation of a coagulopathic state. Therefore, consideration of more targeted interventions including glucocorticoid or anticytokine therapy may be necessary in select cases and requires further study. Notably, in this study we could not correlate coinciding measures of CAR T expansion in patients with bleeding events, which should be included in future investigations. To better elucidate the specific mechanisms of CAR T-related coagulopathy, future studies should also assess coagulation factor levels and related substances, as well as the natural inhibitors of coagulation: protein-C and -S, tissue factor pathway inhibitor (TFPI), and antithrombin III (AT-III). Measurement of vitamin K–dependent factor levels or PIVKA-II (protein induced in the absence of vitamin K) may provide suitable measures of vitamin K status34 and help individualize management strategies in patients receiving broad-spectrum antibiotics or with other conditions predisposing to vitamin K deficiency. Manual review of peripheral smears may also elucidate better understanding of microangiopathy processes and should be considered routinely in the first 4 weeks after CAR T therapy. Because of the number of bleeding and thrombosis events, multivariable analysis in our study required the use of a penalized regression model that identified baseline thrombocytopenia as a sole characteristic associated with subsequent bleeding events in our cohort. Future studies with a larger cohort of patients may identify additional covariates predictive of bleeding. Our finding may also imply that steady-state platelet-endothelial interactions are essential for maintaining the integrity of the vascular bed after CAR T therapy.35 Although speculative, finding would justify the clinical observations in our study and provide a novel biologic rationale for both bleeding events and perhaps blood-brain barrier disruption after CAR T therapy.
The majority of thrombotic complications occurred within 60 days after CAR T infusion, including 3 events at the time of disease progression. In part because of the limited number of thrombotic events, no characteristics or risk factors for thrombosis could be identified in our study. We observed a trend toward increased thrombotic events in patients with a history of thrombosis (12% vs 5%), which was not statistically significant. However, our study may not be adequately powered for this analysis. We also note that older age, active malignancy, chemotherapy, and hospitalization are all known risk factors for thrombosis, and it is possible that the thrombotic events we observed were attributable more to concurrent risk factors in our study population than to direct toxicity of CAR T therapy. Further investigation with larger study cohorts are necessary to distinguish unique contributions of CAR T toxicity from other risk factors for thrombosis. We did observe that anticoagulation can be safely administered in these patients without subsequent bleeding complications. Ten percent of the patients in our cohort were also safely maintained on anticoagulation for indications that predated CAR T, although temporary discontinuation was required in some patients due to thrombocytopenia. Based on these findings and those of other groups13 we believe anticoagulation can be safely given with close monitoring and the appropriate indications in patients who develop venous thromboembolism after CAR T therapy.
CRS and ICANS are unique toxicities observed with CAR T therapy and associated inflammation can hypothetically trigger both a coagulopathic and thrombotic state. Bleeding and thrombosis were not associated with CRS in our study cohort. However, only 3 patients (2.4%) developed grade ≥3 CRS, which limited our ability to analyze this association. Interestingly, patients with high-grade ICANS (grades 3-4) had a higher incidence of bleeding and thrombosis after CAR T therapy. Moreover, patients with high-grade ICANS more frequently showed a pattern of consumptive coagulopathy. Multiple groups have shown high-grade ICANS to be associated with DIC and other markers of endothelial activation including increased von Willebrand factor, ratios of ANG2 to angiopoietin 1, and autopsy findings suggestive of endothelial activation and fibrin deposition.12,36,37 We find it intriguing that Holtzman et al also found that elevated baseline fibrinogen levels were associated with high-grade ICANS in a cohort of patients with large B-cell lymphoma treated with axi-cel.38 This, together with our observation of a more profound descent in serum fibrinogen in patients with high-grade ICANS, suggests that the magnitude of these changes may be a sensitive marker for endothelial dysfunction associated with ICANS. Both clinical and preclinical models suggest that the development of ICANS is dependent on monocyte-lineage cells and monocyte-derived cytokines.39-41 Accordingly, patients with high-grade ICANS have also been shown to have similar cytokine profiles and increased myeloid cells in the cerebral spinal fluid, compared with those without ICANS.42 Strati et al observed lower peak peripheral blood monocyte counts in patients with high-grade ICANS, supporting the notion that extravasation of monocytes into tissue including the central nervous system could have a role in the biology of ICANS.43 Likewise, in our cohort, we observed lower circulating monocyte counts, not only in patients with higher grade ICANS but also in those with bleeding events, and a trend toward the same finding in those with thrombosis (P = .08).
We acknowledge that a major limitation of our analysis is its retrospective design, and future prospective studies are needed to test the hypotheses generated from our findings. Our cohort includes 2 constructs CAR T constructs, which may make our findings more widely applicable. Our results were similar when our analysis was limited to patients with LBCL treated with axi-cel.
In summary, we observed both bleeding (9.4%) and thrombotic (6.3%) events in the first 3 months after CAR T therapy, with bleeding limited to the first month in our cohort. Patients with low baseline platelet counts and even those developing high-grade ICANS may be at highest risk of bleeding events and should be monitored closely. Our observations suggest that systemic coagulopathy/DIC-like picture coincides with bleeding complications after CAR T therapy and is associated with high-grade ICANS. Risk factors for bleeding and thrombotic complications should be studied prospectively to develop risk-assessment models and clinical guidelines for management of bleeding and thrombosis (including prophylaxis) during CAR T therapy.
Acknowledgments
The authors thank the staff and faculty of the Stanford University Blood and Marrow Transplantation Program and the Stanford Center for Cancer Cell Therapy.
This work was supported by National Institutes of Health (NIH), National Cancer Institute grant 2P01CA049605-29A1; Stanford BMT/CCT T32 Training Grant 5T32HL007952-19 (A.J.); Stanford Clinical and Translational Science KL2 Career Development Award program, Award Number KL2 TR003143 (S.S.); and NCI grant K08CA248968 (M.F.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Authorship
Contribution: A.J., J.C., M.F., and S.S. designed the study and interpreted the data; A.J., J.C., J.B., J.S, M.F., and S.S. contributed to data collection; A.J., M.F., S.S., and J.T. analyzed the data and created the figures; A.J., M.F., and S.S. wrote the manuscript; and all authors contributed to data analysis, writing and revision of the manuscript, and approval of the final version.
Conflict-of-interest disclosure: S.S. has consulted for Janssen and has received research funding from Magenta Therapeutics. L.M. has received research funding from Adaptive and Servier and has consulted for Amgen. A.J. is a current equity holder in BioEclipse Therapeutics, a private company; has consulted KUUR Therapeutics and Amgen; is a current equity holder in Magenta Therapeutics, a publicly traded company; and has received honoraria from UpToDate. J.S. is a current equity holder in and a member of the board of directors of Jasper Therapeutics, a private company. E.M. has received research funding from Orca Bio. P.S. has received research funding from Kite (a Gilead Company) and Orca Bio. A.R. has received research funding from Pharmacyclics. C.M. has consulted for and is an equity holder in Apricity Health, Lyell Immunopharma, and Syncopation Life Sciences, all private companies; and has consulted for NeoImmune Tech, Nektar Therapeutics, BMS, Immatics, and Glaxo-Smith-Kline. D.M. has consulted for and has received travel support and research funding from Adaptive Biotech, Juno-Celgene-Bristol-Myers Squibb, and Novartis; has consulted for, received travel support and research funding from, and holds membership on the board of directors of Kite-Gilead; has received research funding from Allogene Therapeutics Inc; has consulted for, received travel support from, and holds patents and royalties from Pharmacyclics; has consulted for and received travel support from Janssen; and has received research funding from Miltenyi Biotec. T.L. has served on the speakers’ bureau of Kite-Gilead. The remaining authors declare no competing financial interests.
Correspondence: Surbhi Sidana, Stanford University, 300 Pasteur Dr, Room H0101, Stanford, CA 94305-5623; e-mail: surbhi.sidana@stanford.edu; and Matthew Frank, Stanford University, 300 Pasteur Dr, Room H0101, Stanford, CA 94305-5623; e-mail: mjfrank@stanford.edu.

