

Key Points
Forty-five percent of patients with a t-MN after RT had chromosome 5 and/or 7 abnormalities.
Patients that developed a t-MN with a recurring translocation had a significantly shorter latency period between RT and development of t-MN.
Abstract
Therapy-related myeloid neoplasms (t-MNs) are a late complication of cytotoxic therapy and are defined as a distinct entity by the World Health Organization. Although the link between chemotherapy exposure and risk of subsequent t-MN is well described, the association between radiation monotherapy (RT) and t-MN risk is less definitive. We analyzed 109 consecutive patients who developed t-MNs after RT and describe latencies, cytogenetic profile, mutation analyses, and clinical outcomes. The most common cytogenetic abnormality was a clonal abnormality in chromosome 5 and/or 7, which was present in 45% of patients. The median latency from RT to t-MN diagnosis was 6.5 years, with the shortest latency in patients with balanced translocations. One-year overall survival (OS) was 52% and 5-year OS was 22% for the entire cohort. Patients with chromosome 5 and/or 7 abnormalities experienced worse 1-year OS (37%) and 5-year OS (2%) compared with other cytogenetic groups (P < .0001). Sixteen patients underwent net-generation sequencing; ASXL1 and TET2 were the most commonly mutated genes (n = 4). In addition, 17 patients underwent germline variant testing and 3 carried pathogenic or likely pathogenic germline variants. In conclusion, patients with t-MN after RT monotherapy have increased frequencies of chromosome 5 and/or 7 abnormalities, which are associated with poor OS. In addition, pathogenic germline variants may be common in patients with t-MN after RT monotherapy.

Introduction
Therapy-related myeloid neoplasms (t-MNs) are a late complication of cytotoxic therapy (chemotherapy and/or ionizing radiation) encompassing cases of myelodysplastic syndrome (MDS), MDS/myeloproliferative neoplasm overlap syndrome, and acute myeloid leukemia (AML) that occur at any time after the treatment.1,2 The World Health Organization has included t-MNs as a distinct entity because of shared clinical, morphologic, and cytogenetic features and to provide insight into the mutagenic effects of known iatrogenic and, by extension, environmental carcinogens on human hematopoietic cells. Multiple mechanisms have been proposed for the development of t-MNs, including DNA damage leading to point mutations or loss of tumor suppressor genes, direct induction of fusion oncogenes, induction of genomic instability causing haploinsufficiency and/or development of complex cytogenetic karyotypes and complex aberrations, selection of preexisting malignant hematopoietic cell clones, induction or exacerbation of a permissive (eg, inflammatory) bone marrow microenvironment, and inherited cancer susceptibility.3-8 Pathogenic TP53 mutations and other genes commonly mutated in clonal hematopoiesis may be present before cytotoxic therapy and subsequently drive the emergence of a t-MN clone.9-11 Of note, whole genome sequencing studies have demonstrated similar rates of single-nucleotide variants and transversions in de novo AML and t-MNs, suggesting that cytotoxic therapy may not be responsible for irreparable genome-wide DNA damage. Rather, cytotoxic therapy may select for therapy-resistant hematopoietic stem/progenitor cells that already have a pathogenic mutation such as TP53.9 Alternatively, environmental and iatrogenic exposures may lead to similar genetic changes.2 Commonly described latency periods, the interval between initial exposure and diagnosis of t-MN, include a 2- to 3-year latency in patients presenting with recurring chromosomal rearrangements after exposure to topoisomerase II inhibitors and a 5- to 7-year latency after exposure to alkylating agents or radiation therapy (RT).12,13 The risk of t-MN after treatment with chemotherapy is independent of the underlying primary tumor type. A recent Surveillance, Epidemiology, and End Results (SEER) Database analysis by Morton et al13 demonstrated a statistically significant increased risk for development of t-MN on receipt of chemotherapy by patients with 22 of 23 analyzed solid tumor histologies.
Although overall survival (OS) of patients with t-MNs is poor, prognosis is influenced primarily by patient characteristics and cytogenetic and molecular profile of the disease.14 An earlier series of 306 patients with t-MNs by Smith et al15 demonstrated a median OS of 8 months for the entire cohort with worse OS in individuals with chromosome 5 and/or 7 abnormalities. Compared with de novo AML, patients with t-MNs are more likely to have chromosomal abnormalities in chromosome 5 and/or 7.15,16 In addition, patients with t-MN have higher rates of pathogenic mutations in KIT, TP53, SF3B1, and JAK2 compared with patients with de novo myeloid neoplasms.16,17 Germline mutations also have a role in t-MN leukemogenesis. In a previous analysis from our center, 21% of patients with breast cancer who subsequently developed a t-MN carried a germline mutation in a cancer susceptibility gene.8
The role of chemotherapy exposure and t-MN risk is well described, but the association between therapeutic RT as a sole modality and subsequent t-MN occurrence has been controversial.18-20 Long-term follow-up of atomic bomb survivors in Japan has established radiation as a risk factor for MN.21-23 Environmental exposure to nuclear power plant accidents has also raised concern.24 More recently, radiation exposure from abdominopelvic computed tomography imaging has been associated with development of MN.25 It has been argued that high radiation doses are lethal to hematopoietic stem cells, and thus DNA damage would not be perpetuated into daughter cells. However, hematopoietic stem cells located within the penumbra region of radiation at the edges of the fields are exposed to much lower doses of ionizing radiation.26,27 Epidemiologic studies of individuals living in proximity to nuclear sites and exposed to low doses of ionizing radiation have demonstrated higher frequencies of point mutations in RUNX1 and loss of heterozygosity at chromosome 5q and 7.28,29
A recent analysis of patients diagnosed with AML or MDS after exposure to RT alone reported cytogenetic features and outcomes more similar to patients with de novo AML.30 This report prompted the question whether AML or MDS after RT monotherapy should be considered therapy related. In reply, our group previously reported clonal cytogenetic findings and clinical outcomes from 71 patients with t-MN after RT monotherapy and found the same poor OS and increased rates of chromosome 5 and/or 7 abnormalities as observed in t-MN after chemotherapy alone.31,32 Here, we analyzed an expanded series of 109 consecutive patients studied at our institution who developed t-MN after RT only. We report latencies, cytogenetic changes, and survival outcomes. When available, we also analyzed somatic and germline mutations in patients in our series. We conclude that our patients with t-MNs experience clinical and molecular changes consistent with t-MN syndromes.
Methods
We included any patient with a t-MN without restricting eligibility based on minimum or maximum latency. Undoubtedly, this allows some patients who may have developed AML/MDS independently of known exposure to fall into this diagnostic category, particularly those with very long latencies. We excluded patients with a primary myeloid malignancy. We excluded patients who received cytotoxic chemotherapy before the t-MN diagnosis and included only those who had received RT monotherapy.Cases of t-MN after RT monotherapy from 1971 to 2020 were identified from the Therapy-Related Leukemia registry at the University of Chicago. We included patients who received radioactive iodine I131 because this agent is a gamma emitter.33 We excluded patients who received only large numbers of diagnostic radiographs or computed tomography scans. Clinical data were abstracted by individual chart review, including the indication for RT, under an institutional review board–approved protocol (UC #4186). The study was conducted accordance with the Declaration of Helsinki. Cytogenetic abnormalities were characterized in metaphase cells and classified as follows: normal karyotype, chromosome 5 abnormalities, chromosome 7 abnormalities, chromosome 5 and 7 abnormalities, recurring translocations, trisomy 8, and other clonal cytogenetic abnormalities. We used the definition for a clone described by the International System for Human Cytogenomic Nomenclature.34 Latency analyses were stratified by RT indication and cytogenetic abnormality. OS analyses were analyzed by cytogenetic abnormality and intensity of antileukemia therapy. Analyses of latency and OS were performed via the Kaplan-Meier method and generated using R software.35
A subset of patients in this cohort underwent next-generation sequencing (NGS) of marrow or blood samples using the University of Chicago Medicine (UCM)-OncoHeme, UCM-OncoScreen, and UCM-OncoPlus platforms. OncoHeme and OncoScreen are amplicon-based NGS panels using MiSeq instruments, whereas UCM-OncoPlus is a 1213 gene hybrid capture NGS panel.36 UCM-OncoPlus data were processed using a custom in-house pipeline, consisting of adapter trimming, alignment to the hg19 version of the human genome, filtering of low mapping quality alignment, and insertion/deletion (indel) realignment. Variant calling was performed after removing PCR duplicates, using a combination of Samtools 0.1.19 and UCM-developed software, Variant Inspector. Average sequencing depth was approximately 800 times, and standard variant and indel calling was performed with variant allelic frequency (VAF) cutoffs of 10% and a minimum variant depth of 50 reads. Mutations with between 5% and 10% VAF were manually reviewed. Manual evaluation of sequencing data was conducted at the pathologists’ discretion for particular mutations of interest present at VAF < 5% using control samples. Nonsynonymous variants were annotated using Alamut Batch 1.3 software (http://www.interactive-biosoftware.com/) and filtered based on their 1000 Genome frequency, coding effect, and location (only exonic variants or those within 6 bp of intron/exon boundaries were considered for this analysis).37 Synonymous variants and common sequencing artifacts were removed.
Germline testing results for this cohort were first available starting in 2011. Patients eligible for germline testing were identified via personal and/or family history. The risks and benefits of germline sequencing were discussed with each patient. For patients who consented to dedicated germline sequencing, a punch biopsy of skin was performed at the time of a bone marrow biopsy or during a dedicated outpatient visit. Skin fibroblasts were cultured and used to extract germline DNA. Germline DNA was sequenced using Clinical Laboratory Improvement Amendments–certified NGS panels at the University of Chicago Human Genetics Clinical Laboratory. All genetic testing results were disclosed by a physician with training in clinical genetics and/or a certified genetic counselor.
Results
Patient characteristics
One hundred nine consecutive patients who developed t-MN after RT monotherapy were identified from 1971 to 2020. Forty-eight patients were diagnosed with t-MDS, whereas 61 patients had t-AML. RT indications are summarized in Table 1. The most common indications were genitourinary (n = 44), breast (n = 31), and gynecologic cancers (n = 14). External beam RT included large portals encompassing areas of active marrow hematopoiesis, such as the pelvis, ribs, and/or sternum, despite appropriate shielding in most cases. Median age at RT was 64 years old (range, 0-83 years old).
RT indications, age at RT exposure, and latencies
| Primary diagnosis | Number of patients | Median age at RT (range, y) | Median latency, y (IQR) |
|---|---|---|---|
| Nonmalignant* | 5 | 21 (7-40) | 6 (3-10) |
| Hematologic cancer† | 4 | 45 (32-51) | 2 (2-4) |
| Solid tumor | |||
| Prostate/testicular | 44 | 69 (41-80) | 6 (3-9) |
| Breast | 31 | 64 (32-83) | 6 (3-9) |
| Gynecologic | 15 | 55 (20-82) | 8 (3-11) |
| Head and neck | 4 | 61 (47-64) | 9 (2-15) |
| Thyroid | 3 | 58 (0-77) | 32 (18-46) |
| Lung | 2 | 66 (58, 73) | 3 |
| Medulloblastoma | 1 | <1 | 45 |
| Total | 109 | 64 (0-83) | 6.5 (3-11) |
| Primary diagnosis | Number of patients | Median age at RT (range, y) | Median latency, y (IQR) |
|---|---|---|---|
| Nonmalignant* | 5 | 21 (7-40) | 6 (3-10) |
| Hematologic cancer† | 4 | 45 (32-51) | 2 (2-4) |
| Solid tumor | |||
| Prostate/testicular | 44 | 69 (41-80) | 6 (3-9) |
| Breast | 31 | 64 (32-83) | 6 (3-9) |
| Gynecologic | 15 | 55 (20-82) | 8 (3-11) |
| Head and neck | 4 | 61 (47-64) | 9 (2-15) |
| Thyroid | 3 | 58 (0-77) | 32 (18-46) |
| Lung | 2 | 66 (58, 73) | 3 |
| Medulloblastoma | 1 | <1 | 45 |
| Total | 109 | 64 (0-83) | 6.5 (3-11) |
Nonmalignant conditions include thyroid pathology (n = 3), hydatidiform mole (n = 1), and acne (n = 1).
Primary hematologic malignancies included Hodgkin lymphoma (n = 3) and non-Hodgkin lymphoma (n = 1).
Cytogenetic abnormalities at t-MN diagnosis are summarized in Table 2. Eighty-three (76%) patients had clonal cytogenetic abnormalities. Forty-five percent of patients had a clonal abnormality involving loss of the long arm of chromosome 5 (22 patients), chromosome 7 (11 patients), or both (16 patients), with or without other abnormalities; 24% had a normal karyotype; 14% had a recurring rearrangement, 10% had trisomy 8; and 7% had other clonal abnormalities. Eighteen patients were noted to have a loss of 17p, which is associated with loss of the TP53 gene. Among the 18 patients with 17p deletion, 17 also had chromosome 5 and/or 7 abnormalities. Of the 43 patients (39%) with complex karyotype, 35 had clonal abnormalities of chromosomes 5 and/or 7. When the cytogenetic profiles were analyzed using European LeukemiaNet 2017 risk stratification,38 6% of patients were classified as favorable risk, 46% as intermediate risk, and 48% as poor risk.
Cytogenetic abnormalities, latencies, and OS of patients with t-MNs after RT monotherapy
| Karyotype | Number of patients (%) | Median latency, y (IQR) | 1-y OS % (95% CI) | 5-y OS % (95% CI) |
|---|---|---|---|---|
| Normal | 26 (24%) | 7.2 (3-10) | 77 (62-95) | 41 (25-66) |
| Clonal abnormalities | ||||
| Abnormal 5, 7, or both* | 49 (45%) | 6.6 (4-11) | 37 (24-53) | 2 (0-15) |
| Abnormal 5 | 22 | 4.4 (3-11) | 36 (21-63) | 5 (1-31) |
| Abnormal 7 | 11 | 6.6 (6-10) | 46 (24-87) | 0 |
| Abnormal 5 and 7 | 16 | 6.5 (5-8) | 31 (15-65) | 0 |
| Recurring translocations | 15 (14%) | 2.3 (2-5) | 55 (34-90) | 55 (34-90) |
| t(8;21) | 1 | 2.1 | 100 | 100 |
| t(21q22) | 2 | 3.7 | 0 | 0 |
| t(15;17) | 4 | 2.2 | 50 | 50 |
| inv(16) | 5 | 2 | 75 | 75 |
| t(16;16) | 1 | 2.3 | 100 | 100 |
| t(11q23.3)/KMT2A | 1 | 7.1 | 0 | 0 |
| inv(3q) | 1 | 35 | 100 | NA |
| Trisomy 8 | 11 (10%) | 6.5 (4-15) | 55 (32-94) | 36 (17-80) |
| Other clonal abnormalities | 8 (7%) | 12.9 (9-45) | 63 (37-100) | 13 (2-78) |
| All cytogenetic analyses | 109 | 6.5 (3-11) | 52 (44-63) | 22 (15-32) |
| Karyotype | Number of patients (%) | Median latency, y (IQR) | 1-y OS % (95% CI) | 5-y OS % (95% CI) |
|---|---|---|---|---|
| Normal | 26 (24%) | 7.2 (3-10) | 77 (62-95) | 41 (25-66) |
| Clonal abnormalities | ||||
| Abnormal 5, 7, or both* | 49 (45%) | 6.6 (4-11) | 37 (24-53) | 2 (0-15) |
| Abnormal 5 | 22 | 4.4 (3-11) | 36 (21-63) | 5 (1-31) |
| Abnormal 7 | 11 | 6.6 (6-10) | 46 (24-87) | 0 |
| Abnormal 5 and 7 | 16 | 6.5 (5-8) | 31 (15-65) | 0 |
| Recurring translocations | 15 (14%) | 2.3 (2-5) | 55 (34-90) | 55 (34-90) |
| t(8;21) | 1 | 2.1 | 100 | 100 |
| t(21q22) | 2 | 3.7 | 0 | 0 |
| t(15;17) | 4 | 2.2 | 50 | 50 |
| inv(16) | 5 | 2 | 75 | 75 |
| t(16;16) | 1 | 2.3 | 100 | 100 |
| t(11q23.3)/KMT2A | 1 | 7.1 | 0 | 0 |
| inv(3q) | 1 | 35 | 100 | NA |
| Trisomy 8 | 11 (10%) | 6.5 (4-15) | 55 (32-94) | 36 (17-80) |
| Other clonal abnormalities | 8 (7%) | 12.9 (9-45) | 63 (37-100) | 13 (2-78) |
| All cytogenetic analyses | 109 | 6.5 (3-11) | 52 (44-63) | 22 (15-32) |
NA, not available.
Seventeen patients in this category also had loss of 17p.
Treatment was individualized according to the patient's wishes, fitness, cytogenetic features, and persistence of primary cancer. Treatment course was available for 84 patients; 26 (30%) received intensive remission induction with cytarabine plus an anthracycline, 35 (41%) received hypomethylating agent-based therapy, 18 (21%) received only supportive care, and 5 (6%) received other therapies. In the latter group were several patients with therapy-related acute promyelocytic leukemia (t-APL) who received all-trans retinoic acid with or without arsenic trioxide. After remission induction, 14 patients (17%) subsequently underwent allogeneic hematopoietic cell transplantation (allo-HCT).
Latency period and survival outcomes
Median latency from RT to t-MN diagnosis was 6.5 years (interquartile range [IQR], 3-11 years). The shortest latency was observed in patients with recurring translocations (n = 15), with a median latency of 2.3 years (P = .002). Median latency was 7.2 years in patients with a normal karyotype, 6.6 years in patients with chromosome 5 and/or 7 abnormalities, 6.5 years in patients with trisomy 8, and 12.9 years in patients with other abnormalities (Table 2; Figure 1B). Latency was also analyzed by RT indication (Figure 1B). The latency periods were similar across RT indications, suggesting that t-MN was associated with the radiation administered and not the underlying primary disease. The shortest median latency of 2.1 years was observed in the 4 patients with a primary hematologic malignancy. Median latency by RT indication was 6 years in patients with breast cancer, 5.6 years in patients with prostate or testicular cancer, 7.7 years among patients with gynecologic malignancies, 6.2 years in patients with other solid tumors, and 4.7 years for nonmalignant conditions (n = 5).

Latency time between RT and development of t-MN. (A) Latency time by cytogenetic group. (B) Latency time by primary diagnosis.
Latency time between RT and development of t-MN. (A) Latency time by cytogenetic group. (B) Latency time by primary diagnosis.
Median OS for the cohort was 13 months (IQR, 5-46 months) with 1-year OS of 52% and 5-year OS of 22% (Table 2). Patients with chromosome 5 and/or 7 abnormalities experienced significantly worse 1-year OS (37%; 95% confidence interval [CI], 24%-53%) and 5-year OS (2%; 95% CI, 0%-15%; P < .0001). The OS for patients with chromosome 5 abnormalities, chromosome 7 abnormalities, or both 5 and 7 abnormalities was equally poor (Figure 2A). The median OS of this group was 9 months (IQR, 3-15 months). The 1-year OS was 55% (95% CI, 34%-90%) for patients with recurring translocations, 55% (95% CI, 32%-94%) for patients with trisomy 8, 63% (95% CI, 37%-100%) for patients with other clonal abnormalities, and 77% (95% CI, 62%-95%) for those with a normal karyotype (Table 2; Figure 2A).

OS of patients with t-MN. (A) OS according to cytogenetic subgroups. (B) OS by year of t-MN diagnosis.
OS of patients with t-MN. (A) OS according to cytogenetic subgroups. (B) OS by year of t-MN diagnosis.
Given the evolution of both leukemia-directed therapies and supportive therapies from 1971 to 2020, we also analyzed survival outcomes based on the year of t-MN diagnosis. We divided patients into those diagnosed before 2000 (n = 42) and those from 2000 onward (n = 67). Patients who were diagnosed from 2000 to the present had better survival outcomes with a 1-year OS of 60% (95% CI, 49%-74%) and a 5-year OS of 27% (95% CI, 18%-41%; Figure 2B). Sixty-two percent of patients with a diagnosis of t-MN before 2000 had a chromosome 5 and/or 7 abnormality, whereas 34% of patients diagnosed with t-MN from 2000 to present had a chromosome 5 and/or 7 abnormality.
For the 84 patients with data on initial t-MN therapies, patients receiving supportive therapy alone experienced shorter OS (median, 6 months; IQR, 4-13 months), 1-year OS (29%, 95% CI, 11%-61%), and 5-year OS (18%; 95% CI, 6%-29%). Patients who received intensive remission induction therapy had a 1-year OS of 67% (95% CI, 53%-84%) and 5-year OS of 27% (95% CI, 16%-47%). Patients receiving therapy with a hypomethylating agent had a 1-year OS of 52% (95% CI, 38%-78%) and 5-year OS of 21% (95% CI, 9%-50%). Patients receiving other targeted therapies had a 1-year OS of 40% (95% CI, 14%-100%) and 5-year OS of 20% (95% CI, 3%-100%; Table 3; Figure 3). Fourteen patients (17%) went on to receive an allo-HCT as part of their treatment course. Within this cohort, the 1-year OS was 79% (95% CI, 60%-100%) and the 5-year OS was 47% (95% CI, 27%-84%). Those who did not receive an allo-HCT (n = 70) had a 1-year OS of 49% (95% CI, 38%-62%), and a 5-year OS of 19% (95% CI, 12%-32%; P = .018).
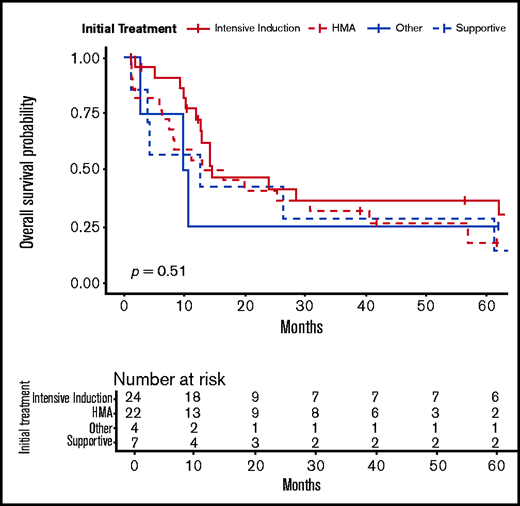
OS according to initial t-MN therapy. HMA, hypomethylating agent (azacitidine or decitabine).
OS according to initial t-MN therapy. HMA, hypomethylating agent (azacitidine or decitabine).
OS of patients by initial t-MN–directed therapy
| Initial therapy | No. patients (%) | 1-y OS % (95% CI) | 5-y OS % (95% CI) |
|---|---|---|---|
| Intensive induction | 38 (45%) | 67 (53-84) | 27 (16-47) |
| HMA | 24 (29%) | 52 (38-78) | 21 (9-50) |
| Other therapy | 5 (6%) | 40 (14-100) | 20 (3-100) |
| Best supportive care | 17 (20%) | 29 (14-61) | 18 (6-49) |
| All therapies | 84 | 54 (44-66) | 24 (16-35) |
| Initial therapy | No. patients (%) | 1-y OS % (95% CI) | 5-y OS % (95% CI) |
|---|---|---|---|
| Intensive induction | 38 (45%) | 67 (53-84) | 27 (16-47) |
| HMA | 24 (29%) | 52 (38-78) | 21 (9-50) |
| Other therapy | 5 (6%) | 40 (14-100) | 20 (3-100) |
| Best supportive care | 17 (20%) | 29 (14-61) | 18 (6-49) |
| All therapies | 84 | 54 (44-66) | 24 (16-35) |
HMA, hypomethylating agent.
Somatic and germline mutations
Somatic NGS data were available for 16 patients at diagnosis of t-MN; these patients were all diagnosed with t-MN in 2015 or later. The most common somatic mutations were ASXL1 (n = 4), TET2 (n = 4), IDH2 (n = 3), U2AF1 (n = 3), TP53 (n = 2), NPM1 (n = 2), FLT3-ITD (n = 2), NRAS (n = 2), STAG2 (n = 2), SRSF2 (n = 2), and CBL (n = 2) (Figure 4; supplemental Table 1). The median number of co-occurring pathogenic mutations was 2 (range, 0-5). Among the 17 patients who had germline testing performed, 3 patients (18%) had a pathogenic or likely pathogenic variant. All 3 patients had an initial diagnosis of prostate cancer and later presented with AML and a normal karyotype. The first patient had a latency of 3.5 years after RT. Although somatic NGS did not demonstrate any pathogenic mutations, germline testing identified a CHEK2 p.Ile157Thr mutation. Of note, the initial somatic NGS panel sent for this patient did not sequence CHEK2. The second patient had a latency of 10 years after RT and somatic NGS showed an ASXL1 p.Glu635Arg mutation. Germline testing identified a DDX41 p.Pro258Leu mutation; DDX41 was not sequenced on the somatic NGS panel. The third patient had a latency of 8 years and did not have somatic NGS at time of t-MN diagnosis. However, germline testing demonstrated a PALB2 p.Arg170Ilefs*14 mutation. Family histories of each patient are summarized in supplemental Table 2.

Discussion
This series of 109 consecutive patients who developed t-MNs after RT monotherapy is, to our knowledge, the largest series specifically focused on this patient subset. We show that t-MNs after RT monotherapy share many of the characteristics of t-MNs that follow cytotoxic chemotherapy. Forty-five percent of patients had a chromosomal abnormality involving chromosome 5 and/or 7 leading to loss of material from the long arms, and this was significantly associated with shorter OS. The incidence of chromosome 5 and/or 7 abnormalities in de novo AML ranges from 10% to 20%,38-40 suggesting enrichment of this cytogenetic abnormality in our RT cohort. The presence of del(5q) or concurrent del(5q) and −7/del(7q) in patients with myeloid malignancies is associated with genomic complexity and poor prognosis.1,2,15,41 In addition, only 24% of patients in our cohort had normal karyotypes, whereas approximately 40% of patients with de novo AML have a normal karyotype.38-40 These frequencies of cytogenetic abnormalities stand in contrast to those reported by Nardi et al,30 who reported a 43% frequency of normal karyotype and only 26% frequency of chromosome 5 and/or 7 abnormalities in their 47 patient cohort with t-MN after RT alone. Of note, we found a higher frequency of chromosome 5 and/or 7 abnormalities in patients diagnosed with t-MN before 2000 in comparison with those diagnosed in 2000 or later, which may reflect the evolution in RT techniques over time. Patients with recurring balanced translocations had an estimated 5-year OS of 55%; this group contained 6 patients with therapy-related core-binding factor AML (t-CBF-AML), and 4 patients with t-APL. Although patients with t-APL have excellent outcomes with all-trans retinoic acid–based therapy, patients with t-CBF-AML have inferior outcomes compared with those with de novo CBF-AML.42,43 Previous studies also demonstrated inferior outcomes in patients with t-AML with favorable cytogenetics compared with de novo AML with favorable cytogenetics.44 This suggests that favorable cytogenetics may not fully ameliorate the poor risk associated with prior cytotoxic therapy in t-MNs.
We found a significantly shorter median latency of 2.3 years in patients who developed recurrent translocations, consistent with findings previously reported by Smith et al15 and others.45 Treatment decisions were based on individual patient characteristics such as age, performance status, cytogenetic features, and persistence of primary malignancy. Patients who received leukemia-directed therapy instead of best supportive care had superior OS. However, other factors beyond cytogenetic features, such as patient fitness, likely influenced this finding. Similarly, individuals who received an allo-HCT had significantly improved OS compared with those who did not, but this does not capture the effect of complete remission rates and patient fitness to undergo an allo-HCT.
Our analysis also included somatic and germline NGS, although the number of patients who underwent either somatic or germline NGS was small. The most frequent pathogenic somatic mutations were in ASXL1 and TET2 (4 each). A median of 2 somatic mutations were present in each patient evaluated. TP53 mutations were noted in 2 (13%) patients; both patients had chromosome 5 and chromosome 7 abnormalities, and 1 also had deletion of 17p. One of the patients with a TP53 mutation underwent germline testing, which did not demonstrate any pathogenic variants. Previous work analyzing the genomic landscape of t-MNs identified ETV6 and EZH2 mutations as being more common in t-MN patients after RT alone, but we did not identify mutations in these 2 genes in any of our patients.17 Eighteen percent (n = 3) of tested patients in this series had pathogenic or likely pathogenic germline mutations. All 3 patients with germline mutations had prostate cancer. Previous work has identified RT as a risk factor for development of t-MN in patients with prostate cancer, but the potential impact of germline variants on t-MN has not been described in that patient population.46 Because allo-HCT is now a standard treatment of t-MN,47 careful genetic screening of related donors may be required.48,49 Furthermore, comprehensive germline testing may offer better insights into the genetic mechanisms that increase the risk for t-MN leukemogenesis.8
Mechanisms leading to t-MN leukemogenesis are multifactorial, with contributions from germline predisposition, DNA damage, and genomic instability increasing the likelihood of complex cytogenetic abnormalities.3-7 In addition to these mechanisms, ionizing radiation disrupts bone marrow stroma and the stem cell microenvironment.50 Subsequent defects in hematopoietic stem cells can be noted after exposure to low-dose radiation.51 A minimum dose of radiation exposure associated with a definitively increased risk of t-MN has not been determined; however, work by Lee et al25 documented an increased risk of myeloid neoplasm after perioperative abdominopelvic computed tomography scans and estimated a mean exposure of 14.7 mGy to the active marrow. Occupational exposures to ionizing radiation, such as nuclear power generation or space travel, may predispose individuals to development and evolution of clonal hematopoiesis, which has been identified as a risk factor for myeloid neoplasm.52,53
Our study has limitations, because it is a retrospective, single-center analysis spanning several decades during which RT techniques and treatment of myeloid malignancies have evolved. Specific data pertaining to radiation doses and fields were not uniformly available. Additional studies focused on understanding how the germline genetic and perhaps epigenetic milieu and treatment-level factors such as treatment type (chemotherapy, RT, or combined modality therapy) impact t-MN leukemogenesis are warranted. In addition, newer cancer therapies such as poly (ADP-ribose) polymerase inhibitors, peptide receptor radionucleotide therapy, and hematopoietic growth factors have been associated with the development of t-MN.54-56 Analysis of the cytogenetic profile, molecular features, and germline predisposition syndromes associated with these t-MNs will be necessary to understand and hopefully minimize the risk conferred by treatment with these DNA-damaging agents.
In summary, our data provide additional evidence to associate RT with the emergence of myeloid neoplasms. The OS of t-MN after RT alone is impacted by cytogenetic factors, as we observed higher rates of chromosome 5 and/or 7 abnormalities in t-MNs compared with concurrent cohorts of de novo AML. Our patients with a recurring translocation had a significantly shorter latency between RT and t-MN diagnosis, consistent with previous studies. In addition, our study suggests that pathogenic germline variants may be common in patients with t-MN (18%). Comprehensive germline testing in the evaluation of t-MNs may also shed further light onto the mechanisms of predisposition to t-MN development.
Acknowledgments
The authors thank Richard L. Lin and Smita S. Joshi for participation in earlier phases of this project.
Authorship
Contribution: A.A.P., A.E.R., and R.A.L. designed the research, analyzed data, and wrote the manuscript. M.W.D., H.W., L.A.G., and M.M.L.B. analyzed data, reviewed the manuscript, and provided edits.
Conflict-of-interest disclosure: L.A.G. serves on an advisory committee for Invitae, Inc. and receives royalties from UpToDate. M.M.L.B. serves on the Board of Directors for Varian Medical Systems and receives royalties from UpToDate. R.A.L. has acted as a consultant or advisor to Amgen, Ariad/Takeda, Astellas, Celgene/BMS, CVS/Caremark, Epizyme, MorphoSys, and Novartis; has received institutional clinical research support from Novartis, Astellas, Celgene, Cellectis, Daiichi Sankyo, Forty Seven/Gilead, Novartis, and Rafael Pharmaceuticals; and has received royalties from UpToDate. The remaining authors declare no conflicting financial interests.
Correspondence: Anand A. Patel, Section of Hematology/Oncology, Department of Medicine, The University of Chicago Medicine, 5841 S Maryland Ave, MC 2115, Chicago, IL 60637; e-mail: anand.patel@uchospitals.edu.

