

Key Points
Systemic bevacizumab was effective in treating severe bleeding in a patient with Heyde syndrome (aortic stenosis with GI angiodysplasia).
As in this case, bevacizumab may enable safe antiplatelet therapy needed for aortic valve replacement in bleeding Heyde syndrome patients.

Abstract
Heyde syndrome, the co-occurrence of aortic stenosis and bleeding gastrointestinal (GI) angiodysplasia, is managed with aortic valve replacement. However, severe bleeding and anemia can preclude safe use of the antiplatelet or anticoagulant therapy required for this intervention. We present a case of the novel and successful treatment of severe, refractory bleeding and transfusion dependence with antiangiogenic therapy in a patient with Heyde syndrome. After systemic bevacizumab was initiated, the patient achieved durable hemostasis with normalization of hemoglobin and liberation from red cell transfusion and dependence on iron infusion; aspirin therapy was successfully initiated where it had previously failed. This durable hemostasis facilitated her subsequent successful transcatheter aortic valve replacement. Plasma vascular endothelial growth factor levels, which were monitored during therapy, paradoxically rose after bevacizumab was initiated but normalized after it was discontinued. Given the angiogenic dysregulation of Heyde syndrome, systemic bevacizumab may be an effective and safe targeted therapy for managing refractory GI bleeding, which thereby facilitates antiplatelet therapy and aortic valve replacement in these challenging cases. Additional investigation into the therapeutic role of inhibiting angiogenesis as a hemostatic modality in Heyde syndrome is warranted.
Introduction
Heyde syndrome is characterized by bleeding from gastrointestinal (GI) angiodysplasias in patients with aortic stenosis. Angiogenic dysregulation due to loss of high molecular weight von Willebrand factor multimers is believed to stimulate angiodysplasia formation in Heyde syndrome.1,2 Management of Heyde syndrome involves replacement of the stenotic aortic valve, but valve replacement requires antiplatelet and/or anticoagulant therapy, which may be contraindicated in the setting of severe bleeding. Here we report the novel use of systemic bevacizumab, a recombinant, humanized monoclonal IgG1 antibody directed against vascular endothelial growth factor (VEGF, also known as VEGF-A), to successfully treat refractory transfusion-dependent anemia secondary to GI angiodysplasias in a patient with severe aortic stenosis. Antiangiogenic therapy enabled safe use of antiplatelet therapy that had previously failed and thus allowed for subsequent successful transcatheter aortic valve replacement (TAVR).
Case description
A 72-year-old woman with a history of mild anemia secondary to small bowel angiodysplasias was in her usual state of health when she developed progressive dyspnea on exertion and chronic, persistent melena. Laboratory workup demonstrated a newly decreased hemoglobin of 6.4 g/dL from her baseline of 10.0 g/dL. Cardiac evaluation with transthoracic echocardiogram demonstrated a mean aortic valve gradient of 62 mmHg, peak aortic valve velocity of 4.6 m/s, and aortic valve area of 0.8 cm2, which are consistent with severe aortic stenosis. Her clinical picture was thought to represent Heyde syndrome, characterized by the co-occurrence of aortic stenosis with bleeding GI angiodysplasias (with acquired von Willebrand syndrome additionally contributing to bleeding in some patients).3
Her anemia was remarkable for a requirement of 23 total packed red blood cell (pRBC) transfusions and 8 infusions of iron sucrose (a total of 2000 mg elemental iron) over 12 months to maintain her hemoglobin in the range of 7 to 8 g/dL (Figure 1). Ongoing GI blood loss was confirmed by numerous fecal occult blood tests and direct visualization of bleeding lesions on multiple endoscopies; therapeutic interventions on endoscopy included repeated treatment with argon plasma coagulation and bipolar cautery. Of note, the patient did not meet any of the Curaçao diagnostic criteria for hereditary hemorrhagic telangiectasia, a condition also associated with GI angiodysplasias; she did not have epistaxis, mucocutaneous telangiectasias, visceral arteriovenous malformations within organs, or a family history of these complications. She continued to have persistent severe anemia despite these local hemostatic interventions, aggressive hematologic support with blood and iron, and 10 months of darbepoetin alfa therapy (200 to 400 μg once every 2 weeks), with a hemoglobin nadir of 5.2 g/dL (Figure 1). She was eventually evaluated for TAVR, and there was an initial concern that she would not tolerate the antiplatelet therapy (aspirin) necessary for transcatheter intervention because of her ongoing severe chronic bleeding and anemia. She was cautiously trialed on aspirin 81 mg once per day when her hemoglobin seemed to transiently improve, but she immediately developed worsening bleeding while receiving aspirin and required 4 units of pRBCs and 1530 mg elemental iron over a 3-week period; this necessitated prompt discontinuation of aspirin. Over the 10 months that followed the aspirin trial, standard systemic therapies to control bleeding failed, including tranexamic acid 1300 mg 3 times per day and octreotide 50 μg 3 times per day. Her bleeding continued to worsen despite these interventions, and she required an additional 35 units of pRBCs to be transfused over this time; these events prompted consideration of a referral for hospice or palliative care.
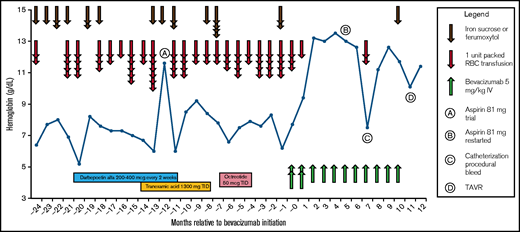
Hemoglobin, hematologic support, and bleeding-directed systemic therapies. The patient required multiple pRBC transfusions on almost a monthly basis to maintain her hemoglobin despite multiple attempted therapies. She was trialed on aspirin therapy at month −12 but continued to have refractory GI bleeding. After starting treatment with bevacizumab 5 mg/kg at time 0, she became transfusion independent and successfully tolerated aspirin, which allowed for her TAVR. TID, 3 times per day.
Hemoglobin, hematologic support, and bleeding-directed systemic therapies. The patient required multiple pRBC transfusions on almost a monthly basis to maintain her hemoglobin despite multiple attempted therapies. She was trialed on aspirin therapy at month −12 but continued to have refractory GI bleeding. After starting treatment with bevacizumab 5 mg/kg at time 0, she became transfusion independent and successfully tolerated aspirin, which allowed for her TAVR. TID, 3 times per day.
Methods
Given the recognition that angiodysplasia formation in Heyde syndrome may be secondary to dysregulated angiogenesis and the lack of any further standard treatment options, systemic bevacizumab therapy was initiated at a dose of 5 mg/kg once every 2 weeks for 2 months (induction) followed by 5 mg/kg once per month (maintenance). Given the nature of this therapy, plasma VEGF-A levels were monitored over the course of treatment by using an assay developed and performed by Mayo Clinic Laboratories (Rochester, MN).
Results and discussion
Within 1 month of initiating treatment with bevacizumab, the patient’s hemoglobin returned to normal for the first time in 5 years (Figure 1), coinciding with a complete normalization of her stools. Four months after bevacizumab treatment was initiated, a second trial of aspirin 81 mg once per day was started, and the patient’s hemoglobin was maintained in the normal range without any significant decline or evidence of GI bleeding. Except for 1 previously scheduled pRBC transfusion administered shortly after the first infusion of bevacizumab, no further pRBC transfusions were required in the 6 months after bevacizumab therapy was started. Bevacizumab was well-tolerated, with no recognizable treatment-emergent adverse events and no occurrence of hypertension, proteinuria, or thrombosis. VEGF-A levels increased during bevacizumab treatment (Figure 2), but they normalized after bevacizumab was discontinued. Seven months after bevacizumab treatment was initiated, the patient experienced a procedural bleeding complication during arterial access for cardiac catheterization for TAVR planning that resulted in hemoglobin drop and pRBC transfusion, but her hemoglobin rapidly normalized afterward. Ten months after bevacizumab initiation, bevacizumab was held, and she underwent successful TAVR 6 weeks later without any need for pRBC transfusion both peri-procedurally and onward. Her hemoglobin has been stable without any evidence of recurrent bleeding or further need for bevacizumab therapy after TAVR.
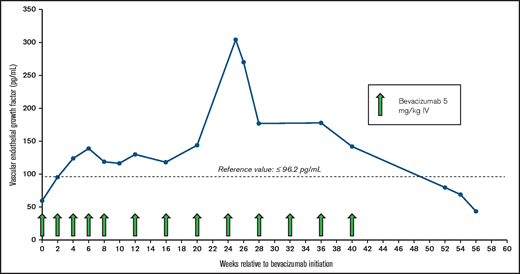
Plasma VEGF levels and bevacizumab treatment course. Plasma VEGF-A levels increased during treatment and normalized after bevacizumab was discontinued. Reference value, ≤96.2 pg/mL. IV, intravenous.
Plasma VEGF levels and bevacizumab treatment course. Plasma VEGF-A levels increased during treatment and normalized after bevacizumab was discontinued. Reference value, ≤96.2 pg/mL. IV, intravenous.
We report a novel case of a patient with longstanding chronic bleeding from GI angiodysplasia (and consequent severe iron deficiency anemia) in the setting of severe aortic stenosis (so-called Heyde syndrome) who was successfully treated with systemic antiangiogenic therapy. Bleeding in Heyde syndrome can be highly morbid or even fatal, and severe anemia concurrent with severe aortic stenosis can be debilitating. Resolution of angiodysplasia and achievement of durable hemostasis in Heyde syndrome often requires aortic valve intervention,4 which may present a clinical dilemma because aortic valve intervention requires tolerance of antiplatelet and/or anticoagulant therapy. Angiodysplasias in Heyde syndrome are thought to occur as a result of dysregulated angiogenesis from the loss of high molecular weight von Willebrand factor multimers (secondary to shear forces from the stenotic aortic valve).1,2 Antiangiogenic intervention could target this critical step in the pathophysiology of angiodysplasia formation. The loss of high molecular weight von Willebrand multimers may also be severe enough to result in a clinically significant acquired von Willebrand syndrome in some patients, resulting in even more severe bleeding manifestations. Unfortunately, we do not know whether our patient had a change in von Willebrand factor testing with bevacizumab therapy because this testing was not performed. This lack of testing is one limitation of our case description, although the association of bleeding GI angiodysplasias with severe aortic stenosis in conjunction with the patient’s stable hemoglobin after withdrawal of bevacizumab after TAVR clearly supports the diagnosis of Heyde syndrome.
Bleeding angiodysplasias as a persistent driver of anemia may be even more problematic because baseline anemia has been shown to be associated with worse outcomes after TAVR, including increased long-term mortality, lack of functional improvement, and higher rate and duration of hospitalizations.5 As a result, the significance of pre-TAVR anemia is an important but often underappreciated consideration in optimizing outcomes for AVR, both transcatheter and surgical. Antiplatelet therapy, at least in the form of low-dose aspirin, is provided for nearly all patients who are scheduled for TAVR.6 Our patient, who required 67 units of pRBCs and 19 iron infusions to maintain a minimally acceptable hemoglobin over the span of 2 years before her eventual TAVR, demonstrates how extreme manifestations of GI bleeding in patients with aortic stenosis may preclude the safe use of antiplatelet therapy, which would effectively prevent consideration of receiving a TAVR.
Given the pathophysiology of angiodysplasia in Heyde syndrome, the failure of all standard treatment approaches, and the effectiveness observed with antiangiogenic therapy to treat GI telangiectasias in hereditary hemorrhagic telangiectasia (an autosomal dominant rare bleeding disorder characterized by disordered angiogenesis),7-11 we offered our patient systemic bevacizumab as a last-resort treatment option. We dosed bevacizumab according to the treatment protocols developed for patients with hereditary hemorrhagic telangiectasia (HHT),12,13 which uses a lower dose than the US Food and Drug Administration–approved oncologic indications. Although systemic bevacizumab is associated with a number of morbid adverse events (such as venous thromboembolism, bowel perforation, and bleeding manifestations) when administered alongside multiagent cytotoxic chemotherapy in patients with cancer, no clear relationship has been observed with any of those complications when bevacizumab was used as a single agent in patients with HHT and no underlying malignancy.7 In our patient, systemic bevacizumab was immediately and strikingly effective for halting bleeding and resolving severe anemia, which enabled safe initiation of antiplatelet therapy and subsequent TAVR. No recurrence of bleeding was observed after TAVR or after bevacizumab was discontinued. Of note, patients with Heyde syndrome sometimes have continued bleeding despite AVR, and systemic bevacizumab could be useful in managing these patients as well.
Recognizing the novel nature of our treatment choice, we monitored plasma levels of VEGF-A before, during, and after treatment with bevacizumab to provide potential insight regarding whether either treatment-related adverse events emerged or bevacizumab resistance developed after an initial response. While neither of these complications occurred, plasma VEGF-A paradoxically increased during treatment and normalized after bevacizumab was discontinued. The assay that we used measures free VEGF-A (not VEGF-A bound to bevacizumab) and seems to be representative of a true increase in concentration of plasma VEGF-A, likely secondary to antibody interference with receptor-mediated endocytosis and protein degradation.14 This phenomenon has been observed in patients with malignancies who receive bevacizumab15-17 and its clinical relevance, if any, is not clear.
In conclusion, we report the novel, rational, and successful use of systemic bevacizumab to manage severe bleeding and anemia in a patient with Heyde syndrome, which facilitated antiplatelet therapy and successful TAVR. Additional investigation into the therapeutic role of inhibiting angiogenesis as a hemostatic modality in Heyde syndrome is warranted.
Acknowledgments
A.B.S. is the recipient of the American Society of Hematology HONORS Award. H.A.-S. is the recipient of the Harvard KL2/Catalyst Medical Research Investigator Training Award and the American Society of Hematology Scholar Award.
Authorship
Contribution: A.B.S. helped draft the manuscript, create the figures, and critically revise the intellectual content of the manuscript; R.S., N.M.G., R.W., and R.S.K. helped critically revise the intellectual content of the manuscript; H.A.-S. conceived and designed the study, helped draft the manuscript, and helped critically revise the intellectual content of the manuscript; and all authors provided final approval of the manuscript.
Conflict-of-interest disclosure: H.A.-S. served as a consultant for Agios, Dova, Rigel, Argenx, Sobi, Moderna and Novartis and received research funding from Agios, Dova, and Amgen. The remaining authors declare no competing financial interests.
Correspondence: Hanny Al-Samkari, Division of Hematology, Massachusetts General Hospital, Harvard Medical School, Zero Emerson Place, Suite 118, Room 112, Boston, MA 02114; e-mail: hal-samkari@mgh.harvard.edu.

