

Key Points
PB-DLBCL is characterized by a GCB phenotype, a centrocyte-like GEP pattern, GCB-associated mutational profile, and favorable prognosis.
These features indicate PB-DLBCL as a distinct extranodal DLBCL entity, and its specific mutations offer potential for targeted therapies.

Abstract
Primary bone diffuse large B-cell lymphoma (PB-DLBCL) is a rare extranodal lymphoma subtype. This retrospective study elucidates the currently unknown genetic background of a large clinically well-annotated cohort of DLBCL with osseous localizations (O-DLBCL), including PB-DLBCL. A total of 103 patients with O-DLBCL were included and compared with 63 (extra)nodal non-osseous (NO)-DLBCLs with germinal center B-cell phenotype (NO-DLBCL-GCB). Cell-of-origin was determined by immunohistochemistry and gene-expression profiling (GEP) using (extended)-NanoString/Lymph2Cx analysis. Mutational profiles were identified with targeted next-generation deep sequencing, including 52 B-cell lymphoma-relevant genes. O-DLBCLs, including 34 PB-DLBCLs, were predominantly classified as GCB phenotype based on immunohistochemistry (74%) and NanoString analysis (88%). Unsupervised hierarchical clustering of an extended-NanoString/Lymph2Cx revealed significantly different GEP clusters for PB-DLBCL as opposed to NO-DLBCL-GCB (P < .001). Expression levels of 23 genes of 2 different targeted GEP panels indicated a centrocyte-like phenotype for PB-DLBCL, whereas NO-DLBCL-GCB exhibited a centroblast-like constitution. PB-DLBCL had significantly more frequent mutations in four GCB-associated genes (ie, B2M, EZH2, IRF8, TNFRSF14) compared with NO-DLBCL-GCB (P = .031, P = .010, P = .047, and P = .003, respectively). PB-DLBCL, with its corresponding specific mutational profile, was significantly associated with a superior survival compared with equivalent Ann Arbor limited-stage I/II NO-DLBCL-GCB (P = .016). This study is the first to show that PB-DLBCL is characterized by a GCB phenotype, with a centrocyte-like GEP pattern and a GCB-associated mutational profile (both involved in immune surveillance) and a favorable prognosis. These novel biology-associated features provide evidence that PB-DLBCL represents a distinct extranodal DLBCL entity, and its specific mutational landscape offers potential for targeted therapies (eg, EZH2 inhibitors).
Introduction
The World Health Organization (WHO) Classification of Soft Tissue and Bone recognizes primary bone lymphoma as a specific lymphoma entity, which is primarily represented by diffuse large B-cell lymphoma (DLBCL).1 Primary bone DLBCL (PB-DLBCL) is a rare DLBCL subtype, with a relative young median age at diagnosis (55 years)2 and a favorable 5-year overall survival (mean OS, 82%).2-9 Most patients present with symptoms of pain, bone fractures, localized swelling, or suspected periprosthetic joint infection.10-13 Patients ’ physical performance can be affected because weight-bearing bones are commonly involved (eg, femur, spine, pelvis).2,6,13
Between studies, reported clinical characteristics and survival rates are diverse due to a lack of strict (anatomical) definitions and consequent proper classification of DLBCL with osseous involvement (O-DLBCL). As such, the WHO classification1 and Messina et al2 distinguish 3 different subentities: PB-DLBCL, with a single bone lesion with or without regional involvement of lymph nodes; polyostotic-DLBCL, with multifocal disease in a single bone or multiple affected bones only; and disseminated-DLBCL, with ≥1 bone lesion(s) and ≥1 (extra) nodal localization(s). These O-DLBCL subentities illustrate patient outcomes, with a superior survival for PB-DLBCL and polyostotic-DLBCL compared with that for disseminated-DLBCL.
Only a few small retrospective cohort studies have investigated the clinicopathologic characteristics of O-DLBCL. Examining cell-of-origin (COO) with immunohistochemistry (IHC) by using the Hans algorithm,14 these studies identified a predominantly germinal center B-cell (GCB) phenotype in ∼60% of O-DLBCL (n = 269 cases, pooled data from 10 studies).5,13,15-22 Based on gene-expression profiling (GEP), this finding was confirmed by Li et al,8 describing a GCB phenotype in 90% (n = 155). Nonetheless, a comprehensive molecular characterization of O-DLBCL is currently lacking.
To our knowledge, only 2 studies report genetic data explicitly collected from O-DLBCL. First, a lack of MYD88 L265P hotspot mutation was observed in O-DLBCLs (n = 15).20 Second, applying a limited targeted next-generation sequencing (tNGS) panel, activating mutations in NOTCH1 and KRAS were identified in PB-DLBCL (n = 1).23 Due to the limited number of studies, relatively small patient cohorts, and absence of comprehensive genetic analyses, knowledge is lacking regarding the genetic constitution of O-DLBCL. This is caused by the rarity of the disease, the difficulty in obtaining sufficient diagnostic tissue, and the inability to attain proper molecular analysis of bone biopsy specimens, as decalcification procedures lead to acquisition of DNA artifacts and complicate interpretation of sequencing results. Consequently, it is unclear whether the various O-DLBCL subentities reflect a separate molecular entity or rather a heterogeneous disease, as commonly assumed for DLBCL, not otherwise specified.24-28
Since the introduction of tNGS, evidence of genetic heterogeneity associated with histopathologic and clinical features and anatomical localization of DLBCL, not otherwise specified has increased. Therefore, the revised (2016) WHO Classification of Tumors of Hematopoietic and Lymphoid Tissues29 recognizes extranodal DLBCL with specific anatomy as separate entities, such as intravascular large B-cell lymphoma, primary cutaneous DLBCL, primary cutaneous DLBCL–leg type, and primary DLBCL of the central nervous system (PCNSL), commonly representing an activated B-cell (ABC) phenotype.30-36 Following this paradigm and because of their specific disease presentation and clinical behavior, we hypothesized that PB-DLBCL contains unique molecular characteristics. To address this theory, our study presents the first comprehensive GEP and targeted deep-sequencing analyses in a well-annotated and relatively large cohort of PB-DLBCL.
Methods
Patient characteristics
This retrospective study investigated 103 cases of O-DLBCLs for which sufficient tumor DNA was available and that were not included in our previous studies.12,19,37-41 Patients were selected through a search of pathology surveys that reported osseous involvement and were diagnosed between 2002 and 2020 at the Leiden University Medical Center (LUMC; n = 48), Amsterdam University Medical Center, location AMC (n = 11), Erasmus MC Cancer Institute (n = 7), or affiliated nonacademic hospitals (n = 37). As an expert center for tumors of soft tissue and bone, the LUMC contribution was enriched for O-DLBCLs. Figure 1A presents an overview of included cases.
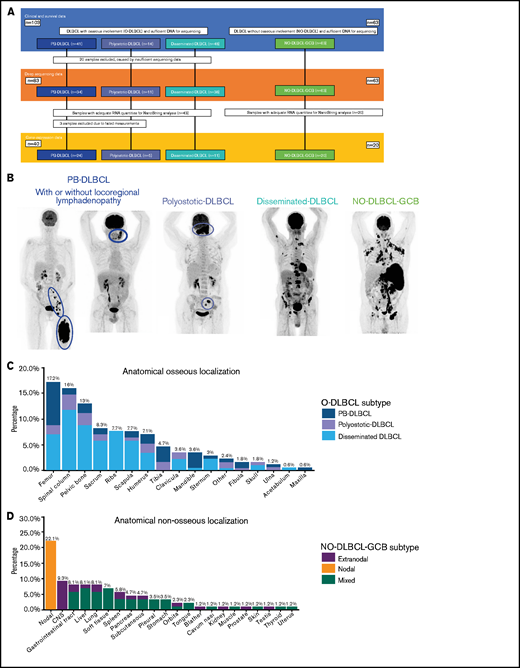
Overview of included O-DLBCL and NO-DLBCL cohorts and subentities with specific anatomical localizations. (A) Flowchart of included and analyzed O-DLBCL and NO-DLBCL subentities. A total of 103 DLBCL cases with osseous involvement were subdivided into three O-DLBCL stages, with PB-DLBCL (n = 41), polyostotic-DLBCL (n = 14), and disseminated-DLBCL (n = 48). Of these, 20 cases failed tNGS quality controls (insufficient DNA or high number of deamination variants), and 83 cases attained appropriate sequencing results. In addition, 63 NO-DLBCL-GCB cases were included as a comparator. Furthermore, 63 samples with adequate RNA were sent for NanoString analysis, of which 3 failed analysis. In total, 24 PB-DLBCL, 5 polyostotic-DLBCL, 11 disseminated-DLBCL, and 20 NO-DLBCL-GCB cases were successfully analyzed with the NanoString platform. (B) Radiologic imaging of International Extranodal Lymphoma Study Group staging system with 3 anatomically defined stages: PB-DLBCL, with a single bone lesion with or without regional involvement of lymph nodes; polyostotic lymphoma (polyostotic-DLBCL), with multifocal disease in a single bone or multiple affected bones; and disseminated lymphoma (disseminated-DLBCL) with ≥1 bone lesion(s) and ≥1 (extra)nodal localization(s).2 NO-GCB-DLBCL was defined as nodal, mixed (nodal and extranodal involvement), or only extranodal localization(s), without any osseous involvement. (C) Frequencies of anatomical osseous localizations identified in all 103 O-DLBCL subentities. Other osseous localizations consisted of one calcaneus, cuneiform, metacarpal III, or talus. (D) Frequencies of anatomical nonosseous localizations of 63 NO-DLBCL-GCBs. CNS, central nervous system.
Overview of included O-DLBCL and NO-DLBCL cohorts and subentities with specific anatomical localizations. (A) Flowchart of included and analyzed O-DLBCL and NO-DLBCL subentities. A total of 103 DLBCL cases with osseous involvement were subdivided into three O-DLBCL stages, with PB-DLBCL (n = 41), polyostotic-DLBCL (n = 14), and disseminated-DLBCL (n = 48). Of these, 20 cases failed tNGS quality controls (insufficient DNA or high number of deamination variants), and 83 cases attained appropriate sequencing results. In addition, 63 NO-DLBCL-GCB cases were included as a comparator. Furthermore, 63 samples with adequate RNA were sent for NanoString analysis, of which 3 failed analysis. In total, 24 PB-DLBCL, 5 polyostotic-DLBCL, 11 disseminated-DLBCL, and 20 NO-DLBCL-GCB cases were successfully analyzed with the NanoString platform. (B) Radiologic imaging of International Extranodal Lymphoma Study Group staging system with 3 anatomically defined stages: PB-DLBCL, with a single bone lesion with or without regional involvement of lymph nodes; polyostotic lymphoma (polyostotic-DLBCL), with multifocal disease in a single bone or multiple affected bones; and disseminated lymphoma (disseminated-DLBCL) with ≥1 bone lesion(s) and ≥1 (extra)nodal localization(s).2 NO-GCB-DLBCL was defined as nodal, mixed (nodal and extranodal involvement), or only extranodal localization(s), without any osseous involvement. (C) Frequencies of anatomical osseous localizations identified in all 103 O-DLBCL subentities. Other osseous localizations consisted of one calcaneus, cuneiform, metacarpal III, or talus. (D) Frequencies of anatomical nonosseous localizations of 63 NO-DLBCL-GCBs. CNS, central nervous system.
Formalin-fixed and paraffin-embedded or fresh frozen tissue samples were obtained during diagnostic procedures (supplemental Table 1). Based on different local standard procedures at the time of initial diagnosis, staging was performed with magnetic resonance imaging, computed tomography imaging, or positron emission tomography/computed tomography scanning and reviewed by a radiologist expert (R.R.) to stratify cases according to WHO definitions (supplemental Table 1).1,2 As a comparator, the study included 63 patients diagnosed between 2006 and 2020 with nonosseous DLBCL as considered by radiologic assessments and a GCB phenotype (NO-DLBCL-GCB) based on the Hans algorithm (Figure 1A). T-cell/histiocyte-rich DLBCL and Burkitt lymphoma were not included. All cases were classified according to Ann Arbor staging and the International Prognostic Index.
This study was performed in accordance with the Dutch Code for Proper Secondary Use of Human Tissue, the local institutional board requirements, and the revised Declaration of Helsinki (2008). It was approved with a waiver of consent by the LUMC’s medical ethics committee (B16.048).
IHC and fluorescence in situ hybridization
Following the latest WHO classification of lymphoid neoplasms,29 IHC and fluorescence in situ hybridization (FISH) analyses were performed (details are given in the supplemental Methods). Briefly, IHC was accomplished with CD10, BCL6, and MUM1 antibodies for COO classification according to the Hans algorithm.14 For O-DLBCLs, MYC, BCL2, and BCL6 rearrangements were analyzed according to FISH, using break-apart probes, as outlined elsewhere.42 NO-DLBCL-GCBs were screened for MYC rearrangements, and if present, BCL2 and BCL6 rearrangements were assessed. Epstein-Barr virus (EBV) status was determined by EBV-encoded RNA in situ hybridization.
Gene expression profiling
GEP was performed with a NanoString system and an extended custom-made probe set, covering 20 genes of the Lymph2Cx assay for COO classification and an additional 219 genes related to DLBCL (supplemental Methods).43-47 For COO classification, raw counts obtained by NanoString gene expression analysis were uploaded at the Lymphoma/Leukemia Molecular Profiling Project Web site for COO categorization (https://llmpp.nih.gov/LSO/LYMPHCX/lymphcx_predict.cgi).48 Technical variation of NanoString nCounter results of each sample was removed by using standardization based on the geometric mean of inherent positive controls in the assay. Next, a principal component analysis was performed as a quality control for identification of possible outliers and potential “batch effects” introduced by NanoString cartridges (supplemental Figure 1). Gene-expression data were normalized by using five Lymph2Cx housekeeping genes, and the resulting data were analyzed with RStudio (R-3.6.3, including packages NanoStringNorm-1.2.1, glmnet-3.0-2, factoextra-1.0.6, ComplexHeatmap-2.2.0, dendextend-1.13.4, ggpubr-0.4.0, and scales-1.1.1). All 234 genes (excluding housekeeping genes) were used to identify GEP clusters within O-DLBCLs and NO-DLBCL-GCB.
Two different assays were used to further subdivide GCB into centroblast or centrocyte B cells. In total, the BAGS(2CLINIC) assays consist of 208 genes, overlapping 26 genes of our custom NanoString panel. Thirteen genes of BAGS(2CLINIC) assays, most distinctive between centroblast and centrocyte B cells, were included for further analysis. In addition, another study recently reported a dark zone/light zone (DZ/LZ) spatial signature consisting of 53 genes, overlapping 11 genes with our panel. Both limited [BAGS(2CLINIC) and DZ/LZ spatial signature] assays, with only MYC overlapping, were used separately to assign centroblast-like or centrocyte-like phenotypes (supplemental Figure 3).49-51
The GEP data reported in this article have been deposited in the Gene Expression Omnibus database (accession number GSE176126) and can also be found in supplemental Table 4.
Targeted next-generation deep sequencing
After microdissection from deparaffinized 10-μm sections (median tumor cells, 70%; range, 20%-90%) (supplemental Table 1), total nucleic acid was isolated with the fully automated Tissue Preparation System (Siemens Healthcare Diagnostics), as previously described.52 For fresh frozen biopsy specimens, DNA was isolated from 25-μm cryosections by using the QIAamp DNA Mini Kit (Qiagen).
The LYMFv1 NGS panel was designed and validated in-house and is an Ion-Torrent–based AmpliSeq panel (Thermo Fisher Scientific; details are provided in the supplemental Methods). The LYMFv1 panel contains 1362 amplicons, subdivided into 2 primer pools, and covers 52 B-cell lymphoma-relevant genes (supplemental Table 2). Briefly, this panel was compiled from a comprehensive review of ∼300 articles (until 2018) on frequencies and clinical relevance of aberrations in B-cell lymphomas. The LYMFv1 panel has an overlap of 73% (33 genes) with a proposed consensus tNGS panel for all mature lymphoid malignancies.53 LYMFv1 libraries were prepared with an Ion Chef System (Thermo Fisher Scientific) or manually. The resulting libraries were sequenced on an Ion Torrent S5-system (Thermo Fisher Scientific). Sequence reads were aligned to the human reference genome (GRCh37/hg19) using TMAP 5.07 software, with default parameters (https://github.com/iontorrent/TS).54 Variants were called by a Torrent Variant Caller. The average read count was 2634 (range, 137-16 001). Supplemental Table 1 lists average read counts per patient. Minimum thresholds for calling variants were ≥100 on-target reads and 10% variant allele frequency. Samples were excluded if deep-sequencing data provided an insufficient number of reads or the transition to transversion ratio was ≥5, indicating excess formalin fixation artifacts. All variants were annotated in the Geneticist Assistant NGS interpretive Workbench (SoftGenetics), into class 1 (benign), class 2 (likely benign), class 3 (unknown significance), class 4 (likely pathogenic), or class 5 (pathogenic).55 Class 4 and 5 variants were designated as pathogenic mutations. Also , class 3 variants of unknown pathogenicity were interpreted as pathogenic mutations, in case of a high Combined Annotation Dependent Depletion–PHRED score (>25) and/or a pathogenic prediction from ≥2 of 4 selected prediction scores (Sift, PolyPhen, the likelihood ratio test, and MutationTaster). Sequencing data obtained for the O-DLBCL and NO-DLBCL-GCB subgroups were mutually compared. In addition, a literature-based cohort of DLBCL-GCB was gathered from 4 large sequencing studies.24-27 Corresponding supplemental Tables (or Figure 524 ) reporting COO subtypes and potential pathogenic variants were used to identify mutational frequencies in DLBCL-GCB cases, collecting a total of 651 DLBCL-GCB cases.
Statistical analysis
Statistical analyses were performed by using RStudio (R-3.6.3, including packages clustertend-1.4, cmprsk-2.2-10, ComplexHeatmap-2.2.0, dendextend-1.13.4, dynpred-0.1.2, factoextra-1.0.6, ggpubr-0.4.0, glmnet-3.0-2, NanoStringNorm-1.2.1, prodlim-2019.11.13, scales-1.1.1, and survival-3.1.11). Hierarchical clustering analysis on GEP data was performed by using Euclidean distance metric and Ward’s minimum variance method for cluster formation. A penalized logistic regression model was applied to identify genes most differentially expressed between PB-DLBCL and NO-DLBCL-GCB.56 This model was based on Elastic Net regression, for which a mixing parameter α of 0.10 was used. Analysis of variance was applied on GEP data of a selected set of 13 genes of the BAGS(2CLINIC) assay and 11 genes of the DZ/LZ spatial signature. The Fisher exact test or Student t test was applied for analyzing categorical or continuous variables among O-DLBCL subgroups and NO-DLBCL-GCB. Progression-free survival (PFS) or OS was defined as date from initial diagnosis to date of progression and/or death by any cause. Patients were administratively censored after 3 years of follow-up or censored at last follow-up when there was no event. The Kaplan-Meier method was used to determine median follow-up time and to construct survival curves, and they were compared with a log-rank test. In case of a statistically significant P value (<.05), corresponding hazard ratios and 95% confidence intervals (CIs) were calculated with a Cox proportional hazards model.
Results
Patient characteristics
O-DLBCL cases were categorized into PB-DLBCL (n = 41), polyostotic-DLBCL (n = 14), and disseminated-DLBCL (n = 48) (Figure 1A-B).17 Table 1 summarizes clinical characteristics of both O-DLBCL and NO-DLBCL-GCB subentities. Individual radiologic assessments and age-related bone localizations are described in the supplemental Results. Figure 1C-D displays exact anatomical non-osseous localizations. Consistent with previous studies, the mean age at diagnosis for PB-DLBCL and polyostotic-DLBCL was (borderline) significantly lower (53 and 50 years) compared with that for disseminated-DLBCL (62 years; P = .020 and P = .068, respectively) and NO-DLBCL-GCB (64 years; P = .003 and P = .033).2,6,7 In addition, NO-DLBCL-GCB cases were subdivided into only nodal (n = 19), mixed (nodal and extranodal involvement, n = 28), or solitary extranodal (n = 16) localization. Six extranodal NO-DLBCL-GCB cases were diagnosed with PCNSL, considered as poor-risk advanced disease (Ann Arbor stage IV) and treated with high-dose methotrexate-containing regimens. Most patients (n = 150 [90%]) were treated with curative intent by using rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone (R-CHOP)-based (immune-) polychemotherapy. Five patients died before treatment, and for palliation, 4 patients received local radiotherapy only or rituximab monotherapy. Median follow-up times for patients with O-DLBCL and NO-DLBCL-GCB were 40 and 20 months, respectively.
Patient characteristics of O-DLBCL and NO-DLBCL-GCB
| Characteristic | PB-DLBCL (n = 41) | Polyostotic-DLBCL (n = 14) | Disseminated-DLBCL (n = 48) | Extranodal NO-DLBCL-GCB (n = 16) | Nodal NO-DLBCL-GCB (n = 19) | Mixed NO-DLBCL-GCB (n = 28) | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Male sex | 24 | 59% | 9 | 64% | 32 | 67% | 10 | 63% | 13 | 68% | 18 | 64% |
| Median age (minimum-maximum), y | 54 (18-86) | 56 (13-73) | 63 (30-91) | 65 (46-82) | 67 (35-84) | 63 (44-95) | ||||||
| Ann Arbor stage | ||||||||||||
| I((X)E) | 29 | 71% | 0 | 0% | 0 | 0% | 4 | 25% | 2 | 11% | 0 | 0% |
| II((X)E) | 12 | 29% | 0 | 0% | 3 | 6% | 1 | 6% | 9 | 47% | 8 | 29% |
| III(E/S) | 0 | 0% | 0 | 0% | 3 | 6% | 0 | 0% | 8 | 42% | 2 | 7% |
| IV | 0 | 0% | 14 | 100% | 42 | 88% | 11 | 69% | 0 | 0% | 18 | 64% |
| IPI score | ||||||||||||
| 0-1 | 26 | 63% | 3 | 21% | 7 | 15% | 6 | 38% | 11 | 58% | 8 | 29% |
| 2-5 | 15 | 37% | 11 | 79% | 41 | 85% | 10 | 63% | 8 | 42% | 20 | 71% |
| First-line treatment | ||||||||||||
| R-CHOP +/− adjuvant radiotherapy* | 35 | 85% | 12 | 86% | 42 | 88% | 8 | 50% | 16 | 84% | 23 | 82% |
| Other chemotherapy ± adjuvant radiotherapy† | 4 | 10% | 2 | 14% | 4 | 8% | 0 | 0% | 1 | 5% | 3 | 11% |
| High-dose MTX‡ ± adjuvant radiotherapy | 0 | 0% | 0 | 0% | 0 | 0% | 7 | 44% | 0 | 0% | 0 | 0% |
| Palliative treatment§ | 2 | 5% | 0 | 0% | 2 | 4% | 1 | 6% | 2 | 11% | 2 | 7% |
| Response to first-line treatment | ||||||||||||
| CR | 37 | 90% | 14 | 100% | 36 | 75% | 11 | 69% | 14 | 74% | 18 | 64% |
| Non-CR | 4 | 10% | 0 | 0% | 12 | 25% | 5 | 31% | 5 | 26% | 10 | 36% |
| Median follow-up, mo | 50 | 53 | 37 | 33 | 17 | 22 | ||||||
| Characteristic | PB-DLBCL (n = 41) | Polyostotic-DLBCL (n = 14) | Disseminated-DLBCL (n = 48) | Extranodal NO-DLBCL-GCB (n = 16) | Nodal NO-DLBCL-GCB (n = 19) | Mixed NO-DLBCL-GCB (n = 28) | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Male sex | 24 | 59% | 9 | 64% | 32 | 67% | 10 | 63% | 13 | 68% | 18 | 64% |
| Median age (minimum-maximum), y | 54 (18-86) | 56 (13-73) | 63 (30-91) | 65 (46-82) | 67 (35-84) | 63 (44-95) | ||||||
| Ann Arbor stage | ||||||||||||
| I((X)E) | 29 | 71% | 0 | 0% | 0 | 0% | 4 | 25% | 2 | 11% | 0 | 0% |
| II((X)E) | 12 | 29% | 0 | 0% | 3 | 6% | 1 | 6% | 9 | 47% | 8 | 29% |
| III(E/S) | 0 | 0% | 0 | 0% | 3 | 6% | 0 | 0% | 8 | 42% | 2 | 7% |
| IV | 0 | 0% | 14 | 100% | 42 | 88% | 11 | 69% | 0 | 0% | 18 | 64% |
| IPI score | ||||||||||||
| 0-1 | 26 | 63% | 3 | 21% | 7 | 15% | 6 | 38% | 11 | 58% | 8 | 29% |
| 2-5 | 15 | 37% | 11 | 79% | 41 | 85% | 10 | 63% | 8 | 42% | 20 | 71% |
| First-line treatment | ||||||||||||
| R-CHOP +/− adjuvant radiotherapy* | 35 | 85% | 12 | 86% | 42 | 88% | 8 | 50% | 16 | 84% | 23 | 82% |
| Other chemotherapy ± adjuvant radiotherapy† | 4 | 10% | 2 | 14% | 4 | 8% | 0 | 0% | 1 | 5% | 3 | 11% |
| High-dose MTX‡ ± adjuvant radiotherapy | 0 | 0% | 0 | 0% | 0 | 0% | 7 | 44% | 0 | 0% | 0 | 0% |
| Palliative treatment§ | 2 | 5% | 0 | 0% | 2 | 4% | 1 | 6% | 2 | 11% | 2 | 7% |
| Response to first-line treatment | ||||||||||||
| CR | 37 | 90% | 14 | 100% | 36 | 75% | 11 | 69% | 14 | 74% | 18 | 64% |
| Non-CR | 4 | 10% | 0 | 0% | 12 | 25% | 5 | 31% | 5 | 26% | 10 | 36% |
| Median follow-up, mo | 50 | 53 | 37 | 33 | 17 | 22 | ||||||
CR, complete response; IPI, International Prognostic Index.
R-CHOP (rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone; n = 136) and adjuvant radiotherapy (n = 47).
CHVmP/BV (cyclophosphamide, doxorubicin, teniposide, and prednisone with bleomycin and vincristine at mid-interval; n = 4), CHOP (N = 1), COPADM (cyclophosphamide, vincristine, prednisone, doxorubicin, and methotrexate; n = 1), DA-EPOCH-R (dose-adjusted etoposide, prednisolone, vincristine, cyclophosphamide, doxorubicin, and rituximab; n = 5), PECC (prednisone, etoposide, chlorambucil, and lomustine; n = 1), RCEOP (rituximab, cyclophosphamide, etoposide, vincristine, and prednisone; n = 1), RCVP (rituximab, cyclophosphamide, vincristine, prednisone; n = 1), and adjuvant radiotherapy (n = 9).
MATRIX (high-dose methotrexate [MTX], cytarabine, thiotepa, and rituximab)/BCNU (Carmustine)/autologous stem cell transplantation (n = 1), MBVP (high-dose MTX, BCNU, teniposide, and prednisone)/HD_araC (high-dose ara-cytarabine) (n = 1), or RMP (rituximab, high-dose MTX, and procarbazine; n = 5), and adjuvant radiotherapy (n = 2).
Local radiotherapy (n = 3 ), Rituximab-monotherapy (n = 1), or no treatment (n = 5).
Pathological features
Figure 2 displays morphologic examples and immunohistochemical characteristics of O-DLBCL. According to the Hans classification, a GCB phenotype was identified in 74% of O-DLBCL (70 of 94 patients) (supplemental Table 3). Using NanoString/Lymph2Cx, a GCB phenotype was found in 88% of O-DLBCL cases (35 of 40 patients), an “intermediate/unclassifiable” phenotype in 10% (n = 4), and an ABC phenotype in 2% (n = 1). In addition, NanoString/Lymph2Cx revealed a GCB phenotype in 90% of NO-DLBCL-GCB cases (18 of 20 patients), one ABC phenotype, and one intermediate/unclassifiable phenotype. Overall, the COO concordance between cases with both IHC and NanoString results was 83% (50 of 60 cases).
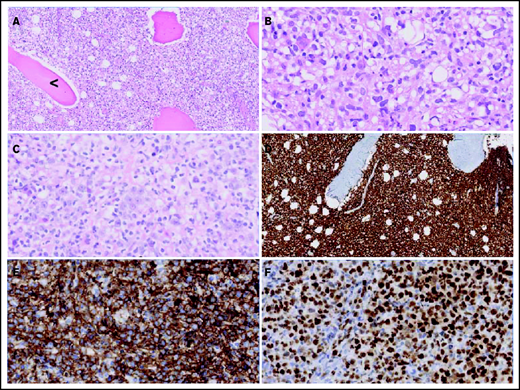
Morphologic and immunohistochemical features of O-DLBCLs. (A) Infiltration of pleomorphic B cells with entrapment of preexisting bone (black arrowhead) in an example of PB-DLBCL. (B) Pleomorphic B cells in a case of PB-DLBCL with large and irregular nuclei with a cleaved, multilobulated appearance and small nucleoli. (C) Pleomorphic B cells in a case of disseminated-DLBCL with large nuclei and prominent large nucleoli with an immunoblastic/plasmablastic appearance. (D) Diffuse staining of CD20 in PB-DLBCL. (E) Diffuse staining of CD10 in an example of PB-DLBCL with a GCB phenotype, according to the Hans algorithm. (F) Strong diffuse staining of MUM1 in an example of disseminated-DLBCL with an ABC phenotype.
Morphologic and immunohistochemical features of O-DLBCLs. (A) Infiltration of pleomorphic B cells with entrapment of preexisting bone (black arrowhead) in an example of PB-DLBCL. (B) Pleomorphic B cells in a case of PB-DLBCL with large and irregular nuclei with a cleaved, multilobulated appearance and small nucleoli. (C) Pleomorphic B cells in a case of disseminated-DLBCL with large nuclei and prominent large nucleoli with an immunoblastic/plasmablastic appearance. (D) Diffuse staining of CD20 in PB-DLBCL. (E) Diffuse staining of CD10 in an example of PB-DLBCL with a GCB phenotype, according to the Hans algorithm. (F) Strong diffuse staining of MUM1 in an example of disseminated-DLBCL with an ABC phenotype.
Fluorescence in situ hybridization
The majority of O-DLBCL cases (83 of 103) were screened for MYC/BCL2/BCL6 rearrangements and EBV status (Figure 3A). Due to technical failures, most likely caused by decalcification of bone material, analysis of all 3 rearrangements failed in 40% (33 of 83 cases). Therefore, only a descriptive analysis was performed. Approximately similar frequencies of rearrangements were identified in polyostotic-DLBCL, disseminated-DLBCL, and NO-DLBCL-GCB, largely consistent with occurrences of DLBCL-GCB cases in the literature.24,27,28,57 Compared with NO-DLBCL-GCB, MYC/BCL2 rearrangements were observed at relatively low frequencies, whereas BCL6 rearrangements were more common (4%, 8%, and 31%, respectively) in PB-DLBCL, indicating that only BCL6 rearrangements seem to be relevant for PB-DLBCL lymphomagenesis. A “double/triple”-hit makeup characteristic for high-grade B-cell lymphoma was observed in ten NO-DLBCL-GCB cases and three disseminated-DLBCL cases but not in PB-DLBCL or polyostotic-DLBCL cases. IHC MYC and BCL2 status for evaluating double expressors are described in the supplemental Results. No O-DLBCL (n = 61) and only three NO-DLBCL-GCB cases were EBV positive. Lack of EBV in these overall GCB subtype DLBCL cases is consistent with previous studies describing the occurrence of EBV-positive DLBCLs mainly (in elderly subjects) with an ABC phenotype.58
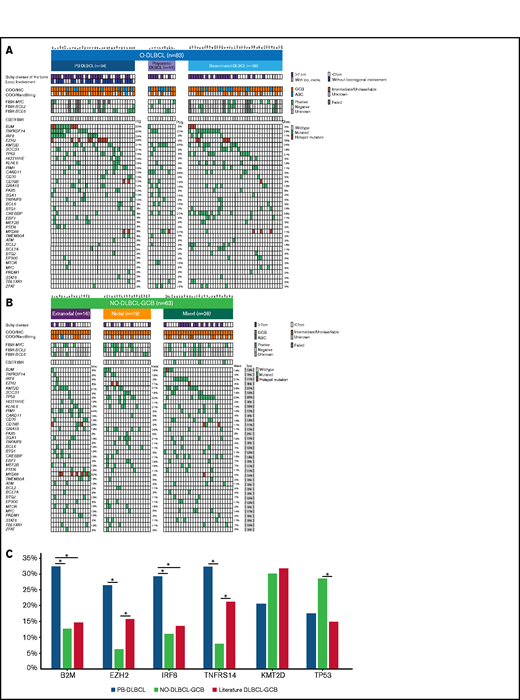
Significant differences in genetic landscapes between PB-DLBCL and NO-DLBCL-GCB. OncoPrint plot of the genetic aberrations and COO of O-DLBCL (A) and NO-DLBCL-GCB (B) subentities. COO phenotype is indicated by blue for ABC, orange for GCB, brown for intermediate (only NanoString), and gray for cases with unknown COO phenotype. Furthermore, a positive ISH (FISH or EBV-encoded small RNA) and a mutation in one of the genes are marked with green. Hotspot mutations are indicated with dark red (B2MM1*, CD79BY196*, EZH2Y646*, and MYD88L265P). (C) Comparison of identified genetic aberrations with high frequencies (≥20%) of PB-DLBCL, NO-DLBCL-GCB, and a pooled literature-based DLBCL-GCB cohort. PB-DLBCL showed a unique genetic profile, with increased frequencies of B2M, EZH2, IRF8, and TNFRSF14, and was significantly different (P = .031, P = .010, P = .047, and P = .003, respectively) compared with NO-DLBCL-GCB harboring high occurrences (although not significant) of KMT2D and TP53 aberrations (P = .347 and P = .325). Except for EZH2 and TNFRSF14 (P = .148 and P = .136), the occurrence of mutations in B2M and IRF8 in our cohort of PB-DLBCL was significantly higher compared with that of the literature-based DLBCL-GCB cohort (P = .012 and P = .020). Careful interpretation is needed, as essential data regarding exact anatomical localizations (eg, osseous involvement was unknown) were lacking for these studies. Significant difference (P < .05) between two groups were marked with asterisks.
Significant differences in genetic landscapes between PB-DLBCL and NO-DLBCL-GCB. OncoPrint plot of the genetic aberrations and COO of O-DLBCL (A) and NO-DLBCL-GCB (B) subentities. COO phenotype is indicated by blue for ABC, orange for GCB, brown for intermediate (only NanoString), and gray for cases with unknown COO phenotype. Furthermore, a positive ISH (FISH or EBV-encoded small RNA) and a mutation in one of the genes are marked with green. Hotspot mutations are indicated with dark red (B2MM1*, CD79BY196*, EZH2Y646*, and MYD88L265P). (C) Comparison of identified genetic aberrations with high frequencies (≥20%) of PB-DLBCL, NO-DLBCL-GCB, and a pooled literature-based DLBCL-GCB cohort. PB-DLBCL showed a unique genetic profile, with increased frequencies of B2M, EZH2, IRF8, and TNFRSF14, and was significantly different (P = .031, P = .010, P = .047, and P = .003, respectively) compared with NO-DLBCL-GCB harboring high occurrences (although not significant) of KMT2D and TP53 aberrations (P = .347 and P = .325). Except for EZH2 and TNFRSF14 (P = .148 and P = .136), the occurrence of mutations in B2M and IRF8 in our cohort of PB-DLBCL was significantly higher compared with that of the literature-based DLBCL-GCB cohort (P = .012 and P = .020). Careful interpretation is needed, as essential data regarding exact anatomical localizations (eg, osseous involvement was unknown) were lacking for these studies. Significant difference (P < .05) between two groups were marked with asterisks.
Gene expression profiling
Because the NanoString material was limited, GEP was performed on 63 randomly selected cases, of which ∼20 ng/μL of RNA was available (Figure 1A; supplemental Table 5). After excluding 3 failed measurements and 2 outliers (supplemental Figure 1), clustering of GEP data was performed on 58 cases, representing 23 PB-DLBCL, 5 polyostotic-DLBCL, 11 disseminated-DLBCL, and 19 NO-DLBCL-GCB. Using both fresh frozen and formalin-fixed, paraffin-embedded tissues for GEP analysis did not affect the identified difference between O-DLBCL and NO-DLBCL-GCB (supplemental Figure 1E). This finding is consistent with previous studies showing a high correlation between GEP data obtained from fresh frozen and formalin-fixed, paraffin-embedded tissues.59-61 Unsupervised hierarchical clustering with GEP data of an extended Lymph2Cx (234 genes) provided 4 different clusters (clusters A-D) (supplemental Figure 2). The most significant difference was found between cluster A, allocating eight PB-DLBCLs and one NO-DLBCL-GCB, and cluster B, with three PB-DLBCLs and 12 NO-DLBCL-GCBs (P < .001). Cluster C was a mixture of O-DLBCL subentities and cluster D an agglomeration of O-DLBCL subtypes and NO-DLBCL-GCBs. Disseminated-DLBCL was observed across all 4 clusters, indicating its heterogeneity and wide variety in disease origins of individual cases.
To further differentiate GEP differences between PB-DLBCL and NO-DLBCL-GCB, a penalized logistic regression model was performed, assigning a significant set of 34 genes differentially expressed between PB-DLBCL and NO-DLBCL-GCB. Unsupervised clustering of these differentially expressed genes generated 3 clusters: A, predominantly PB-DLBCLs; B, an agglomeration of O-DLBCL subtypes and NO-DLBCL-GCB; and C, mainly NO-DLBCL-GCBs (Figure 4). In contrast to NO-DLBCL-GCB, PB-DLBCL displayed significantly increased expression (P < .001) of immune response genes (CTLA4 and CXCL12) and HLA-A, HLA-C, HLA-E, and HLA-F. Elevated expression levels of ARID1A and SMARCA4 (both involved in chromosome organization) and FOXO1 (a centroblast hallmark) were found in NO-DLBCL-GCB compared with PB-DLBCL.62-65
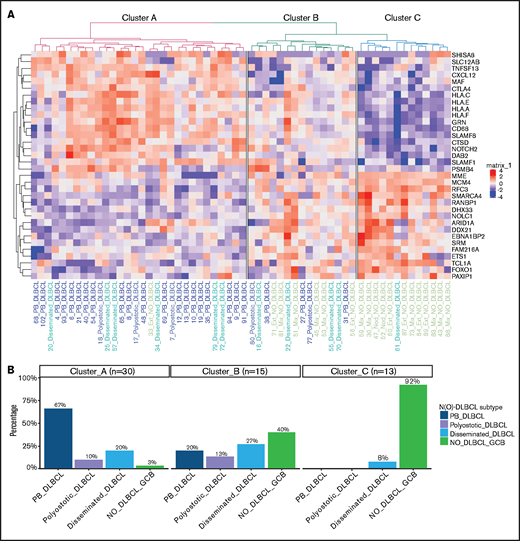
Specific GEP patterns for PB-DLBCL and NO-DLBCL-GCB. (A) A penalized logistic regression model assigned 34 differentially expressed genes between PB-DLBCL and NO-DLBCL-GCB. As shown in the heatmap, unsupervised hierarchical clustering of these differentially expressed genes generated 3 clusters: a cluster with predominant PB-DLBCLs, a cluster with an agglomeration of O-DLBCL subentities and NO-DLBCL-GCB, and a cluster with mainly NO-DLBCL-GCBs. In contrast to NO-DLBCL-GCB, PB-DLBCL exhibited significantly (P < .001) increased expression of immune response genes (CTLA4, CXCL12, HLA-A, HLA-C, HLA-E, and HLA-F). Elevated expressions of ARID1A and SMARCA4 (both involved in chromosome organization) and FOXO1 (a centroblast hallmark) were found in NO-DLBCL-GCBs, as opposed to low expressions in PB-DLBCLs. (B) This bar chart shows the distribution of the PB-DLBCL and NO-DLBCL-GCB subentities across elucidated clusters.
Specific GEP patterns for PB-DLBCL and NO-DLBCL-GCB. (A) A penalized logistic regression model assigned 34 differentially expressed genes between PB-DLBCL and NO-DLBCL-GCB. As shown in the heatmap, unsupervised hierarchical clustering of these differentially expressed genes generated 3 clusters: a cluster with predominant PB-DLBCLs, a cluster with an agglomeration of O-DLBCL subentities and NO-DLBCL-GCB, and a cluster with mainly NO-DLBCL-GCBs. In contrast to NO-DLBCL-GCB, PB-DLBCL exhibited significantly (P < .001) increased expression of immune response genes (CTLA4, CXCL12, HLA-A, HLA-C, HLA-E, and HLA-F). Elevated expressions of ARID1A and SMARCA4 (both involved in chromosome organization) and FOXO1 (a centroblast hallmark) were found in NO-DLBCL-GCBs, as opposed to low expressions in PB-DLBCLs. (B) This bar chart shows the distribution of the PB-DLBCL and NO-DLBCL-GCB subentities across elucidated clusters.
Subsequently, to relatively distinguish between a centroblast-like or centrocyte-like phenotype of PB-DLBCL and NO-DLBCL-GCB cases, both limited BAGS(2CLINIC)-GEP and DZ/LZ spatial signature assays were assessed.49-51 Expression levels of 8 genes (62%) were significantly different between PB-DLBCL and NO-DLBCL-GCB (P ≤ .047) (supplemental Figure 3). PB-DLBCL exhibited significantly higher expression of BCL2A1 and IL6R (centrocyte related), whereas NO-DLBCL-GCB exhibited significantly increased expression for BCL6, MME, MYBL1, FOXO1, SMARCA4, and TCL1A (centroblast related). Applying a limited-DZ/LZ spatial signature, 9 genes showed significantly higher expression (CD3E, CD4, CD8A, CTLA4, FAS, HLA-E, ITGB2, LAG3, and STAT1) for PB-DLBCL compared with NO-DLBCL-GCB (P ≤ .031) (supplemental Figure 3C-D), designating a centrocyte-like phenotype for PB-DLBCL. Despite these limited-BAGS(2CLINIC) and limited-DZ/LZ spatial signature analyses, both independently identified GEP differences indicating a possible centrocyte-like phenotype for PB-DLBCL and a conceivable centroblast-like constitution for NO-DLBCL-GCB, corroborating previous results by Li et al.8
Targeted next-generation deep sequencing
In total, 83 O-DLBCLs and 63 NO-DLBCL-GCBs successfully underwent deep sequencing. For 20 O-DLBCLs, obtained NGS data were of insufficient quality due to DNA artifacts (Figure 1A). Pathogenic variants were identified in 49 genes (Figure 3A; supplemental Table 5), with a median of 4 mutated genes per individual (range, 0-12). Four known “hotspot” mutations were elucidated: loss-of-function B2M p.M1* and CD79B, p.Y196*, and gain-of-function EZH2 p.Y646* and MYD88 p.L265P. In contrast to a prior study, our data revealed low frequencies of MYD88 p.L265P mutation in O-DLBCLs.20 Based on strict anatomical WHO definitions, the 2 most biologically different DLBCL subentities (PB-DLBCL and NO-GCB-DLBCL) were compared to explore potential differences. The mutational profile of PB-DLBCL included frequent mutations (≥25%) in B2M, EZH2Y646*, IRF8, and TNFRSF14 (loss-of-function) and differed significantly from NO-DLBCL-GCB, which was relatively lacking these mutations (P = .031, P = .010, P = .047, and P = .003, respectively) (Figure 3C).
In contrast to PB-DLBCL, high occurrences (≥25%) of KMT2D and TP53 aberrations were observed within NO-DLBCL-GCB (P = .347 and P = .325). In addition to frequent mutations in CREBBP, KMT2D, MYD88, and TNFRSF14, CARD11 was the most commonly mutated gene (36%) in polyostotic-DLBCL and was (borderline) significantly higher compared with PB-DLBCL (P = .085), disseminated-DLBCL (P = .036), or NO-DLBCL-GCB (P = .014), suggesting a biologically distinct subgroup. With frequent mutations in TNFRSF14, KMT2D, or TP53, disseminated-DLBCL exhibited similarities with molecular constitutions of both PB-DLBCL and NO-DLBCL-GCB. In addition, focusing on 19 disseminated-DLBCL cases with bulky osseous disease (Figure 3), comparable mutational profiles as PB-DLBCL were identified with high frequencies of mutations in B2M (16%), EZH2 (26%), IRF8 (21%), and TNFRSF14 (42%), suggesting that this lymphoma originated in bone.
Survival analyses
Consistent with the prognostic importance of International Extranodal Lymphoma Study Group staging,2 PB-DLBCL and polyostotic-DLBCL showed superior PFS/OS (P = .011/P = .042) (Figure 5A-B), compared with disseminated-DLBCL, with 3-year OS rates of 91% (95% CI, 0.83-1.00), 100% (95% CI, 1.00-1.00), and 77% (95% CI, 0.64-0.90), respectively. No significant difference in PFS/OS was observed for extranodal, nodal, and mixed NO-DLBCL-GCB subentities (Figure 5C-D). PB-DLBCL exhibited a significantly superior PFS/OS compared with equivalent Ann Arbor stage I/II NO-DLBCL-GCBs (P = .016/P = .046) (Figure 5E-F). Between PB-DLBCL and NO-DLBCL-GCB, the mutational landscape differed significantly (P = .002), as the majority of PB-DLBCLs (24 of 34) harbored ≥1 mutation in B2M, EZH2, IRF8, and TNFRSF14, compared with a minority of stage I/II NO-DLBCL-GCB (7 of 25) with ≥1 of these specific mutations. No difference was observed in the occurrence of mutations in KMT2D or TP53 between PB-DLBCL (12 of 34) and NO-DLBCL-GCB (12 of 25; P = .423). With respect to Ann Arbor stage III/IV, disseminated-DLBCL and NO-DLBCL-GCB exhibited similar survival outcomes, although polyostotic-DLBCL reported an improved PFS/OS (Figure 5G-H). Besides a prognostic impact for Ann Arbor stage, International Prognostic Index, and age, further univariate survival analyses revealed no remarkable survival differences for patient characteristics, COO, rearrangements, or individual pathogenic variants, presumably due to low patient numbers and the relatively low number of events (supplemental Table 6).
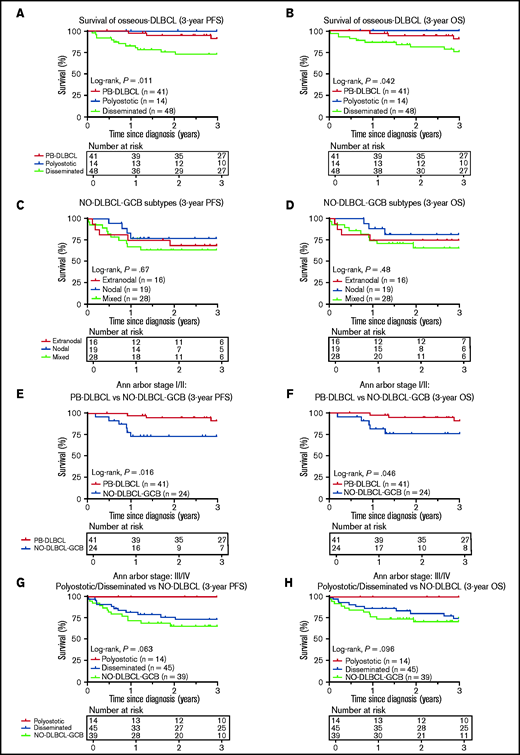
Three-year PFS and OS analysis for O-DLBCL and NO-DLBCL-GCB subentities. (A-B) Consistent with the prognostic importance of International Extranodal Lymphoma Study Group staging, PB-DLBCL and polyostotic-DLBCL displayed a significantly superior PFS and OS, compared with disseminated-DLBCL. (C-D) No significant difference in PFS or OS was shown for the subdivision of NO-DLBCL-GCB into extranodal, nodal, and mixed groups. (E-F) PB-DLBCL elucidated a significantly favorable PFS and OS, compared with equivalent Ann Arbor stage I/II NO-DLBCL-GCBs. (G-H) With respect to Ann Arbor stage III/IV, there was no difference in PFS or OS between disseminated-DLBCL and NO-DLBCL-GCB, although polyostotic-DLBCL showed improved survival.
Three-year PFS and OS analysis for O-DLBCL and NO-DLBCL-GCB subentities. (A-B) Consistent with the prognostic importance of International Extranodal Lymphoma Study Group staging, PB-DLBCL and polyostotic-DLBCL displayed a significantly superior PFS and OS, compared with disseminated-DLBCL. (C-D) No significant difference in PFS or OS was shown for the subdivision of NO-DLBCL-GCB into extranodal, nodal, and mixed groups. (E-F) PB-DLBCL elucidated a significantly favorable PFS and OS, compared with equivalent Ann Arbor stage I/II NO-DLBCL-GCBs. (G-H) With respect to Ann Arbor stage III/IV, there was no difference in PFS or OS between disseminated-DLBCL and NO-DLBCL-GCB, although polyostotic-DLBCL showed improved survival.
Discussion
To our knowledge, this study is the first to provide a comprehensive and integrative evaluation of IHC, GEP, and targeted deep sequencing in a clinically well-annotated and relatively large cohort of patients with O-DLBCL. As previously described,8 IHC/NanoString analysis confirmed a predominant GCB phenotype in O-DLBCL cases, across all subentities. Extended-Lymph2Cx-GEP analysis revealed significantly different clusters for PB-DLBCL specifically targeting immune surveillance genes, in contrast to NO-DLBCL-GCB with a focus on chromosome organization and reduction of p53 activity. Limited-BAGS(2CLINIC) and DZ/LZ spatial signature analysis indicated a centrocyte-like phenotype for PB-DLBCL with a preferential origin in the early LZ of B-cell development, as opposed to a centroblast-like constitution (DZ) for NO-DLBCL-GCB. Intriguingly, the predominant GCB centrocyte-like phenotype in PB-DLBCL was supported by frequent mutations in GCB-associated genes (ie, B2M, EZH2, IRF8, TNFRSF14). In addition, although with a favorable survival in general for DLBCL-GCB, PB-DLBCL with its corresponding specific mutational profile was significantly associated with a superior OS compared with equivalent Ann Arbor limited-stage I/II NO-DLBCL-GCB. Based on our data, we propose a model in which PB-DLBCL can be recognized as a distinct extranodal DLBCL, with a centrocyte-like GCB phenotype, overexpression of immune response genes, and a unique GCB-associated molecular constitution, thereby reflecting a favorable prognosis (Figure 6).
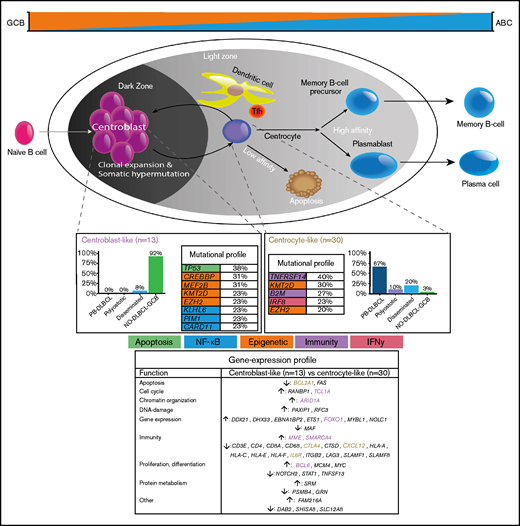
Mechanistical overview of GCB development related to PB-DLBCL and NO-DLBCL-GCB and their identified specific GEP patterns and molecular profiles. As described by Li et al.,8 and corresponding with our GEP analysis showing an increased expression of BCL2A1 and IL6R, PB-DLBCL preferentially originated in the GC early LZ of B-cell development, indicating a centrocyte-like phenotype. The predominant centrocyte-like GCB phenotype in PB-DLBCL was subsequently supported by frequently mutated GCB-associated genes, such as B2M, EZH2, IRF8, and TNFRSF14, culminating in superior survival. In addition, PB-DLBCL exhibited significantly (P < .001) increased expression of immune response genes (CTLA4 and CXCL12), and together with frequent mutations in B2M and TNFRSF14, they are important for immune surveillance, suggesting that evasion from immune surveillance is crucial for PB-DLBCL to survive in their osseous environment. In contrast, upregulation of BCL6, MME, MYBL1, SMARCA4, and TCL1A suggested a centroblast-like constitution for NO-DLBCL-GCB. Accordingly, high expression of FOXO1, a centroblast hallmark and imperative for sustaining the GC DZ,62-65,69 was specifically identified in NO-DLBCL-GCB. Furthermore, elevated expression levels of ARID1A and SMARCA4 (both involved in chromosome organization) were found in NO-DLBCL-GCB. Together with frequent mutations in genes involved in epigenetics (CREBBP and MEF2B) and TP53 mutations, this indicates that, unlike immune evasion in PB-DLBCL, survival of NO-DLBCL-GCB is critically dependent on deregulation of chromosome organization and reduction of p53 activity. Our results thus emphasize that PB-DLBCL can be recognized as a distinct extranodal DLBCL, with a GCB-centrocyte-like phenotype, a specific GEP pattern, and a unique GCB-associated molecular constitution, reflecting favorable prognosis. Purple color indicates genes related to a centroblast-like phenotype, whereas brown-colored genes are related to a centrocyte-like phenotype.
Mechanistical overview of GCB development related to PB-DLBCL and NO-DLBCL-GCB and their identified specific GEP patterns and molecular profiles. As described by Li et al.,8 and corresponding with our GEP analysis showing an increased expression of BCL2A1 and IL6R, PB-DLBCL preferentially originated in the GC early LZ of B-cell development, indicating a centrocyte-like phenotype. The predominant centrocyte-like GCB phenotype in PB-DLBCL was subsequently supported by frequently mutated GCB-associated genes, such as B2M, EZH2, IRF8, and TNFRSF14, culminating in superior survival. In addition, PB-DLBCL exhibited significantly (P < .001) increased expression of immune response genes (CTLA4 and CXCL12), and together with frequent mutations in B2M and TNFRSF14, they are important for immune surveillance, suggesting that evasion from immune surveillance is crucial for PB-DLBCL to survive in their osseous environment. In contrast, upregulation of BCL6, MME, MYBL1, SMARCA4, and TCL1A suggested a centroblast-like constitution for NO-DLBCL-GCB. Accordingly, high expression of FOXO1, a centroblast hallmark and imperative for sustaining the GC DZ,62-65,69 was specifically identified in NO-DLBCL-GCB. Furthermore, elevated expression levels of ARID1A and SMARCA4 (both involved in chromosome organization) were found in NO-DLBCL-GCB. Together with frequent mutations in genes involved in epigenetics (CREBBP and MEF2B) and TP53 mutations, this indicates that, unlike immune evasion in PB-DLBCL, survival of NO-DLBCL-GCB is critically dependent on deregulation of chromosome organization and reduction of p53 activity. Our results thus emphasize that PB-DLBCL can be recognized as a distinct extranodal DLBCL, with a GCB-centrocyte-like phenotype, a specific GEP pattern, and a unique GCB-associated molecular constitution, reflecting favorable prognosis. Purple color indicates genes related to a centroblast-like phenotype, whereas brown-colored genes are related to a centrocyte-like phenotype.
Controversial O-DLBCL definitions complicate a meaningful comparison between individual studies, including small numbers of O-DLBCL cases (n = 4-63) and varying frequencies (25%-86%) of IHC-based GCB phenotypes.5,13,15-21 ,66 Using the Affymetrix GeneChip/BAGS2CLINIC assay, Li et al8 reported a GCB phenotype in 90% of O-DLBCLs (n = 155) and a centrocyte-like phenotype in a small subgroup (n = 11). Likewise, our extended-NanoString/Lymph2Cx-GEP showed significantly different GEP clusters for PB-DLBCL and NO-DLBCL-GCB. Also, limited-BAGS(2CLINIC) and DZ/LZ spatial signature analysis indicated a centrocyte-like phenotype for PB-DLBCL and a centroblast-like constitution for NO-DLBCL-GCB.49-51 In PB-DLBCL, Li et al.8 reported upregulation of major histocompatibility complex class I, extracellular matrix and adhesion, and tumor suppressor genes and downregulation of pro-oncogenes, compared with NO-DLBCL-GCB. Furthermore, high expression of genes involved in the immune response (CTLA4 and CXCL12) was identified in PB-DLBCL, and together with frequent mutations in B2M and TNFRSF14, they are important for immune surveillance. This suggests that evasion from immune surveillance is crucial for PB-DLBCL to survive in their osseous environment.62,63,65,67-69 In contrast, NO-DLBCL-GCB exhibited higher expression of ARID1A and SMARCA4 (chromosome organization through SWI/SNF complex), and both target TP53 (DNA damage response) and CDKN1A (cell cycle inhibitor).64,65,69 The frequent mutations in genes involved in epigenetics (CREBBP and MEF2B) and TP53 mutations indicate that, unlike immune evasion in PB-DLBCL, survival of NO-DLBCL-GCB is critically dependent on deregulation of chromosome organization and reduction of p53 activity.65,69 Increased expression of BCL2A1 and IL6R indicated a centrocyte-like phenotype for PB-DLBCL. Upregulation of BCL6, MME, MYBL1, SMARCA4, and TCL1A suggested a centroblast-like constitution for NO-DLBCL-GCB. Lastly, high expression of FOXO1, a centroblast hallmark and imperative for sustaining the GC DZ, was specifically found in NO-DLBCL-GCB, thereby supporting a centroblast-like phenotype for NO-DLBCL-GCB, as opposed to low/average FOXO1 expression in PB-DLBCL.
Comprehensive reviews by Pasqualucci65 and Mlynarczyk et al.69 independently provide an integral insight into the development of GCB lymphomas. Following these established pathogenic principles, the frequently mutated GCB-associated genes, B2M, EZH2, IRF8, and TNFRSF14 (≥1 present in 68% of PB-DLBCLs), are likely to play a crucial role in GCB lymphomagenesis, as elucidated by our data. Frequent occurrence of mutations in chromatin modifiers and immunomodulators (e.g. EZH2 and TNFRSF14) was observed, and although there are similarities with follicular lymphoma, other genetic abnormalities such as BCL2 translocations were less common in PB-DLBCL (2 of 25). Approximately one-quarter of PB-DLBCL pertained a gain-of-function EZH2 hotspot mutation (Y646*), which abrogates the terminal B-cell differentiation and cell cycle control.70 EZH2 acts as an important GC regulator like BCL6, through silencing of genes by tri-methylation of lysine-27 of histone-3 within the PRC2 complex. As such, EZH2 Y646* hyper-represses CKDN1A and IRF4, increasing proliferation and preventing differentiation toward an activated B cell.65 Consequently, compared with EZH2 wild-type, EZH2-mutated DLBCL seems to be susceptible to tazemetostat (EZH2 inhibitor).69,71 Moreover, B2M loss-of-function, EZH2 gain-of-function aberrations, and downregulation of major histocompatibility complex class I/II will lead to a successful evasion of immune surveillance mechanisms.65,69,72,73 TNFRSF14 loss-of-function mutations are associated with B- and T-lymphocyte attenuator downregulation, thereby initiating a B-cell autonomous activation and lymphoma-supportive microenvironment.69,74,75 Finally, IRF8 is a member of the interferon family of transcription factors, regulating immune response through BCL6 activation. However, an IRF8-driven phenotype alone is insufficient for lymphomagenesis because a second genetic hit is required.76,77 This is consistent with our findings indicating that 57% (12 of 21) of mutated IRF8 cases were accompanied by ≥1 B2M, EZH2, and/or TNFRSF14 abnormalities.
Four NGS studies investigated the mutational landscape of large DLBCL cohorts and also reported on COO (Affymetrix, IHC, and/or NanoString), allowing direct comparison of mutation frequencies in COO-stratified subgroups vs our results in O-DLBCL.24-27 These studies included 96, 60, 331, and 164 DLBCL-GCB cases, respectively. This pooled literature-based DLBCL-GCB cohort (n = 651) yields mutation frequencies for B2M, EZH2, IRF8, KMT2D, TNFRSF14, and TP53 of 15%, 16%, 14%, 32%, 21%, and 15%, respectively (Figure 3C). Except for EZH2 and TNFRSF14 (P = .148 and P = .136), the occurrence of mutations in B2M and IRF8 in our cohort of PB-DLBCL was significantly higher compared with the literature-based DLBCL-GCB cohort (P = .012, and P = .020). Because essential data regarding exact anatomical localizations (with osseous involvement unknown) were lacking for these studies, and control for confounding factors was not possible, it could be assumed that a proportion of DLBCL-GCBs were PB-DLBCLs. Excluding these cases from this literature-based cohort might increase the significance level of this comparison. Although an independent validation study remains indispensable, this external literature-based assessment strengthens our findings by emphasizing that PB-DLBCL could be recognized as a distinct molecular entity characterized by frequent mutations in B2M, EZH2, IRF8, and TNFRSF14, compared with NO-DLBCL-GCB.
Clustering analyses in the noted NGS studies have independently designated different (and partially overlapping) molecular DLBCL subtypes related to COO, prognosis, and potential therapeutic targets. In the current study, the limited tNGS panel used for sequencing and lack of chromosomal aberrations impaired proper molecular classification of O-DLBCL subtypes. As such, supplemental Table 7 summarizes only a derivative of these clusters related to molecular profiles identified in O-DLBCL subentities. Considering frequent mutations in B2M, EZH2, IRF8, and TNFRSF14, PB-DLBCL could primarily be categorized in “good-risk” clusters (eg, C1, C3, EZB, BN2, BCL2), which corroborates our results that PB-DLBCL is associated with favorable survival. This contrasts with other WHO-recognized extranodal DLBCLs, such as PCNSL, primary cutaneous DLBCL–leg therapy, and intravascular large B-cell lymphoma, which are primarily characterized by ABC phenotypes and inferior survival. Our findings in PB-DLBCL coincide with compelling evidence, illustrating superior OS for DLBCL-GCB compared with DLBCL-ABC.2,30-36
The characterized genetic background of PB-DLBCL does not answer the question of whether a lymphoma originates in the bone, as it is assumed that a GC does not exist in bones, or that circulating lymphoma cells are attracted by locally secreted chemokines. The contrast in mutational profiles between PB-DLBCL and NO-DLBCL-GCB (and even more with opposite extranodal DLBCL-ABCs) requires additional investigation into the coherence of these genetic factors and the resulting specific interactions with its microenvironment. Remarkably, no specific (extranodal) DLBCL-GCB entity has yet been recognized in the WHO Classification of Tumors of Hematopoietic and Lymphoid Tissues, and therefore this study can be used as a reference study for DLBCL-GCB. As stated before,78 our findings (re)affirm the supplementary merit of examining well-annotated homogeneous cohorts and invoke the need for additional in-depth evaluation of extranodal DLBCLs.
This study was limited by a percentage of GEP (7%; n = 3) and tNGS (19%; n = 20) dropouts of the O-DLBCL cohort, illustrating difficulties in molecular analysis on decalcified bone tissue, with no indication that this dropout is selective for certain outcomes. By using IHC as the primary COO classifier, several non-GCB IHC cases that harbor a late GCB phenotype are absent from our comparator NO-DLBCL-GCB cohort. Given the low percentage (9%) of dissimilar COO classification by IHC and NanoString in the original study,48 we anticipate that this may have possibly biased our results but to a limited extent. Moreover, this investigation would have benefited from more extensive GEP measurements (eg, complete BAGS2CLINIC assay or DZ/LZ spatial signature assay) for refinement of COO clustering and comprehensive sequencing data (eg, whole-exome sequencing) to elucidate complete molecular profiles, including copy number alterations or larger structural variations. Nevertheless, these techniques would also have been impeded by our perceived (partially) suboptimal DNA/RNA qualities. Furthermore, GEP analyses focused on the comparison of PB-DLBCL with NO-DLBCL-GCB, and therefore the numbers of polyostotic-DLBCL and disseminated-DLBCL analyzed were underrepresented and require additional research. A sensitivity analysis showed that the inclusion of a relatively small number of high-grade B-cell lymphoma cases did not significantly bias our results (supplemental Results). Multivariate analyses showed that the heterogeneity in age, chemotherapy, or adjuvant radiotherapy did not confound our survival outcomes.
In conclusion, this study is the first to show that PB-DLBCL is characterized by a centrocyte-like GCB phenotype, with a specific GEP pattern and GCB-associated mutational profile (mainly B2M, EZH2, IRF8, and TNFRSF14 mutations), both involved in immune surveillance, and is associated with favorable survival. Consequently, these new biological findings provide evidence that PB-DLBCL can be recognized as a distinct extranodal DLBCL entity and offers potential for the development of targeted therapies (eg, EZH2 inhibitors or other epigenetic-modulating agents69 ) to ultimately improve patients’ survival.
Acknowledgments
The authors thank A. Stolk, D. van Egmond, E. de Winter, J. Neefjes, M. Suleiman Ibramoglu, and S. Somers for their valuable technical assistance. They acknowledge the support provided by Louis M. Staudt’s Laboratory at the National Cancer Institute of the National Institutes of Health for the online analysis of Lymph2Cx raw data for COO characterization.
This study is funded by the Stichting Fonds Oncologie Holland.
Authorship
Contribution: Histopathologic samples were provided by P.M.J., V.T., A.N., I.F.-S.., W.C.E.d.H., P.C.W.H., S.T.P., A.D., J.V.M.G.B., and A.H.G.C.; pathology review was performed by P.M.J., V.T., A.N., I.F.-S., W.C.E.d.H., P.C.W.H., S.T.P., A.D., J.V.M.G.B., and A.H.G.C..; R.A.L.d.G., R.v.E., T.v.W., D.R., F.A.d.G., K.K., and I.B.-d.B. gathered data derived from tNGS and FISH; GEP with the NanoString and Lymph2Cx data analysis was provided by A.v.d.B. and A.D.; R.R. performed radiologic review; clinical data regarding patients with NO-DLBCL from other hospitals were provided by L.t.B., H.L., L.H., F.A.d.G. L.H.B., E.F.M.P., M.N., P.J.L., M.J.K., and H.V.; statistical analyses was performed by R.A.L.d.G., S.B., A.H.G.C., and J.S.P.V.; and R.A.L.d.G., A.H.G.C., and J.S.P.V. wrote the manuscript and all authors approved the final manuscript.
Conflict-of-interest disclosure: M.J.K. has received honoraria/research funding from Kite Pharma, Millennium/Takeda, Mundipharma, Gilead Sciences, Bristol Myers Squibb, Roche, Celgene, Novartis Pharmaceuticals Corporation, and Amgen. P.J.L. has received honoraria/research funding from Takeda, Servier, Genmab, Roche, Genentech, Celgene, Incyte, and Regeneron. The remaining authors declare no competing financial interests.
Correspondence: Joost S.P. Vermaat, Department of Hematology, Leiden University Medical Center, PO Box 9600, 2300 RC Leiden, The Netherlands; e-mail: j.s.p.vermaat@lumc.nl.

