

Key Points
Physiologic levels of extracellular IGFBP7 prolong the surface expression and activation of IGF1R by insulin/IGF in ALL.
Knockdown or antibody neutralization of IGFBP7 decrease ALL progression in vivo.

Abstract
Insulin and insulin-like growth factors (IGFs) are mitogenic and prosurvival factors to many different cell types, including acute lymphoblastic leukemia (ALL). Circulating IGFs are bound by IGF binding proteins (IGFBPs) that regulate their action. IGFBP7 is an IGFBP-related protein (IGFBP-rP) that in contrast to other IGFBPs/IGFBP-rPs features higher affinity for insulin than IGFs and was shown to bind the IGF1 receptor (IGF1R) as well. The role of IGFBP7 in cancer is controversial: on some tumors, it functions as an oncogene, whereas in others, it functions as a tumor suppressor. In childhood ALL, higher IGFBP7 expression levels were associated with worse prognosis. Here we show that IGFBP7 exerts mitogenic and prosurvival autocrine effects on ALL cells that were dependent on insulin/IGF. IGFBP7 knockdown or antibody-mediated neutralization resulted in significant attenuation of ALL cell viability in vitro and leukemia progression in vivo. IGFBP7 was shown to prolong the surface retention of the IGF1R under insulin/IGF1 stimulation, resulting in sustained IGF1R, insulin receptor substrate 1 (IRS-1), protein kinase B (AKT), and extracellular signal-regulated kinase (ERK) phosphorylation. Conversely, the insulin receptor was readily internalized and dephosphorylated on insulin stimulation, despite IGFBP7 addition. The affinity of homodimeric IGF1R for insulin is reportedly >100 times lower than for IGF1. In the presence of IGFBP7, however, 25 ng/mL insulin resulted in IGF1R activation levels equivalent to that of 5 ng/mL IGF1. In conclusion, IGFBP7 plays an oncogenic role in ALL by promoting the perdurance of IGF1R at the cell surface, prolonging insulin/IGF stimulation. Preclinical data demonstrate that IGFBP7 is a valid target for antibody-based therapeutic interventions in ALL.
Introduction
Insulin and insulin-like growth factors (IGF1 and IGF2) are well-known mitogenic and prosurvival factors to many different cell types, including both B-cell precursor (BCP) and T-cell acute lymphoblastic leukemias (ALLs).1 Insulin/IGFs act by binding to receptor tyrosine kinases made of homo- or heterodimeric insulin receptor (INSR) and IGF1 receptor (IGF1R) chains, that recruit and phosphorylate insulin receptor substrate proteins (IRS1-4), thus initiating the downstream activation of the phosphatidylinositol 3-kinase/protein kinase B/mammalian target of rapamycin (PI3K/AKT/mTOR) and Ras/Raf/MAPK signaling pathways.2,3 Circulating IGFs are normally bound by 1 of the 6 IGF binding proteins (IGFBPs) that can both inhibit or potentiate IGF action.4 In ALL, IGFBP1, 3, and 4 were shown to inhibit, whereas IGFBP2, 5, and 6 had no influence on, IGF1-induced proliferation of the NALM16 and RS4;11 cell lines in vitro.5
The IGF-binding domain of IGFBPs is shared by some other extracellular proteins, collectively called IGFBP-related proteins (IGFBP-rPs). IGFBP-rPs bind IGFs with low affinity (100-1000 times lower than IGFBPs 1-6) and have multiple IGF-independent roles; therefore, their physiologic significance in the IGFs system remains undefined.6 However, IGFBP-rP1, best known as IGFBP7, is a special case among IGFBP-rPs or IGFBPs, because it binds insulin with relatively high affinity, although with lower affinity than that exhibited by the INSR.7 In short-term experiments (3 minutes), IGFBP7 was shown to inhibit insulin-stimulated INSR signaling.7 In 72-hour cell proliferation assays, on the contrary, IGFBP7 was shown to enhance the mitogenic activities of IGFs and insulin,8 suggesting that physiologically, it may not compete with the INSR/IGF1R for their ligands but instead may augment the half-life of these growth factors.
We previously reported that ALL cells are the main source of IGFBP7 in the leukemia bone marrow microenvironment. The leukemia-secreted IGFBP7 was shown to stimulate bone marrow stromal cells to produce more asparagine, thus counteracting the effect of the antineoplastic drug l-asparaginase.9 In that study, we also found that the knockdown of IGFBP7 in 2 BCP-ALL cell lines (REH and 697) resulted in reduced proliferation,9 suggesting that IGFBP7 could play an autocrine role as well. Here we characterize the autocrine effects of IGFBP7 in ALL, performing preclinical studies to validate it as a target for therapeutic intervention.
Methods
Cell culture
ALL cell lines were cultured in RPMI-1640 (Cultilab, Campinas, São Paulo, Brazil) with 10% fetal bovine serum (FBS), 50 U/mL penicillin, and 50 μg/mL streptomycin (Cultilab). Cryopreserved mononuclear cells from diagnostic bone marrow samples of children with ALL were thawed, depleted of dead cells by Ficoll-gradient centrifugation, and cultured in AIM-V medium (Thermo Fisher Scientific, Waltham, Massachusetts). The study was approved by the Research Ethics Committee from the State University of Campinas (CAAE: 0014.0.144.146-08 and 0018.0.144.146-08) and was conducted in accordance with the Declaration of Helsinki. Animal experiments were approved by the Animal Experimentation Ethics Committees of the State University of Campinas (CEUA/UNICAMP, protocol 1133/2008) and Centro Infantil Boldrini (CEUA/BOLDRINI, protocol 0006/2020).
Short hairpinRNA knockdown of IGFBP7 and IGF1R
IGFBP7 and IGF1R were downregulated using pLKO.1 MISSION short hairpin RNA (shRNA; NM_001553.1-959s1c1 and NM_001553.1-812s1c1) or the pLKO.1 MISSION LacO shRNA (NM_000875) lentiviral vectors (Sigma-Aldrich, San Luis, Missouri), respectively, and their corresponding nontarget shRNA control vectors, as previously described.9
Cell viability and proliferation assays
Cell viability was analyzed by the 3-(4,5-dimethylthiazol-2-yl)-2,5-dimethyltetrazolium bromide (MTT) assay. IGFBP7 knockdown cells were cultured at 30000 cells per well, in 96-well plates, in RPMI-3% FBS. Antibody neutralization of IGFBP7 was analyzed in 96-well plates at 30000 cells per well in RPMI-10% FBS with 20 µg/mL anti-IGFBP7 (clone C311) or anti–prostate-specific antigen (PSA; Rheabiotech, Campinas, São Paulo, Brazil) control. The effects of insulin (500 ng/mL; Novo Nordisk, Bagsværd, Denmark), IGF1 (200 ng/mL; R&D Systems, Minneapolis, Minnesota), IGF2 (200 ng/mL; R&D Systems), and/or IGFBP7 (100 ng/mL; R&D Systems) on cell lines or primary ALL cells was assessed in 96-well plates at 30 000 cells per well in RPMI-10% FBS or AIM-V, respectively. Proliferation of IGFBP7 or IGF1R knockdown cells was assayed in 24-well plates at 50 000 cells per well in RPMI-3% FBS or RPMI-10% FBS, respectively. Numbers of living cells were estimated in a Countess II Cell Counter (Thermo Fisher Scientific) after trypan blue staining.
Apoptosis assays
Cell lines or primary ALL cells were cultured at 50 000 cells per well in 96-well plates in RPMI-3% FBS or RPMI-10% FBS or AIM-V medium alone or supplemented with insulin (500 ng/mL), IGFBP7 (100 ng/mL or 20 µg/mL), anti-IGFBP7 (clone C311; 20 µg/mL), or anti-PSA (20 µg/mL). After 24 or 48 hours, cells were washed with phosphate-buffered saline (PBS), resuspended in Annexin-V binding buffer (Becton Dickinson, Franklin Lakes, New Jersey), and labeled with fluorescein isothiocyanate (FITC)-conjugated Annexin-V (Immuno Tools, Friesoythe, Germany) for 20 minutes and 7-Aminoactinomycin D (7AAD, 5 μg/mL; Thermo Fisher Scientific) for 3 minutes, at room temperature, and immediately analyzed in a LSR Fortessa cytometer (Becton Dickinson) using the FlowJo Software (Becton Dickinson).
5-bromo-2′-deoxyuridinecell cycle assay
We used the FITC 5-bromo-2′-deoxyuridine (BrdU) Flow Kit (Becton Dickinson). Cells at 50 000 cells per well in 96-well plates were starved 4 hours in serum-free RPMI medium for synchronization and then were cultured in RPMI-10% FBS for 24 hours and labeled for 4 hours with BrdU. Finally, cells were labeled with 7AAD and analyzed in a LSR Fortessa cytometer using the FlowJo software.
Migration assay
Thirty thousand cells were seeded in the upper chamber of transwell culture plate inserts with 5-µm pores (Corning, New York) in RPMI-10% FBS. Stromal cell-derived factor 1 (SDF-1, 100 ng/mL; Sigma-Aldrich) or vehicle was added in the lower chamber to stimulate migration. After 4 hours of incubation, cells in the lower chamber were counted in a LSR Fortessa cytometer using the FlowJo software.
Enzyme-linked immunosorbent assay
Enzyme-linked immunosorbent assay (ELISA) of IGFBP7 in conditioned culture medium was described elsewhere.9 Cell lines or primary ALL cells were cultured in serum-free RPMI or AIM-V medium, respectively, for 4 hours, at 250,000 cells per well, in 48-well plates. Cells were left untreated or treated with insulin (500 ng/mL) and/or IGFBP7 (100 ng/mL) for 15 minutes or 4 hours, pelleted, and lysed in RIPA-like buffer (50 mM Tris-HCl; 150 mM NaCl; 1% NP-40; 0.5% sodium deoxycholate; 0.1% sodium dodecyl sulfate) supplemented with 1% phosphatase inhibitor cocktail I (Sigma-Aldrich), 1% phosphatase inhibitor cocktail II (Sigma-Aldrich), 1% protease inhibitor cocktail (Sigma-Aldrich), and 1 mM phenylmethylsulfonyl fluoride (Sigma-Aldrich). Lysates were analyzed using the PathScan Sandwich ELISA Antibody Pair Kits for Phospho-IRS-1 (panTyr), Phospho-Akt1 (Ser473), Phospho-IGFIRβ (Tyr1131), and Phospho-INSRβ (Tyr1150/1151; Cell Signaling Technology, Danvers, Massachusetts). To quantify IGF1Rβ and INSRβ proteins, we adapted the Sandwich ELISA Antibody Pair Kit protocols. Briefly, plates were coated overnight at 4°C, using 10 μg cell lysate in 100 µL/well. The capture antibodies offered in the PathScan kits were used to detect IGF1Rβ and INSRβ, followed by incubation with a horseradish peroxidase–conjugated anti-mouse immunoglobulin G (IgG; 1:1000, Cell Signaling Technology).
Anti-IGFBP7 monoclonal antibody production
Balb/c mice were immunized with a IGFBP7-derived peptide antigen (sequence 100% homologous in human and mouse) by standard methods.10 Hybridomas were selected by ELISA against the native IGFBP7.9 The detection limit reached by clone C311 in ELISA against the bovine serum albumin (BSA)-conjugated antigen peptide was 0.05 ng (data not shown). Of note, the peptide antigen has no relevant similarity against any other protein as evaluated by BLASTp.
Western blot
Cell lines or primary ALL cells were cultured for 4 hours in serum-free RPMI or AIM-V medium, respectively, and then left untreated or stimulated with insulin (500 ng/mL), IGFBP7 (100 ng/mL), and/or anti-IGFBP7 (clone C311; 20 µg/mL) for 15 minutes or 4 hours and then pelleted and lysed in RIPA-like buffer. Thirty micrograms of protein was electrophoresed in 10% sodium dodecyl sulfate-polyacrylamide gels and electroblotted onto nitrocellulose membranes. Membranes were immunoblotted overnight at 4°C with antibodies (Cell Signaling Technology) against phospho-Akt (Ser473; clone 9271), phospho-p44/42 MAPK (Erk1/2; Thr202/Tyr204; clone 9101), and β-actin (clone 4967) in Tris-buffered saline with Tween 20 (20 mM Tris, 150 mM NaCl, 0.1% Tween-20) with 2% BSA (Sigma-Aldrich). Immunodetection was performed by incubation with horseradish peroxidase–conjugated anti-rabbit IgG (1:5,000; Cell Signaling Technology) in 5% nonfat dry milk in Tris-buffered saline with Tween 20, for 1 hour at room temperature, and developed using the Super Signal West Pico Chemiluminescent Substrate (Thermo Fisher Scientific). Images were acquired with a ChemiDoc equipment (Bio-Rad, Hercules, California).
INSR and IGF1R internalization assay
ALL cells (2.5 × 105) were starved in serum-free medium (RPMI for cell lines and AIM-V for primary cells) for 4 hours and then were left untreated or stimulated with insulin (500 ng/mL) and/or IGFBP7 (100 ng/mL) for 15 minutes or 4 hours. When indicated, the C311 anti-IGFBP7 antibody was added 210 minutes after the beginning of the 4-hour starvation time. After washing with PBS, the surface expression of INSR and IGF1R was analyzed by labeling cells with the anti-hCD220-PE (clone 3B6; Becton Dickinson) and anti-hCD221-BV421 (clone 1H7; Becton Dickinson) antibodies or the corresponding isotype controls (mIgG1k-PE, clone MOPC-21, and mIgG1k-BV421, clone X40; Becton Dickinson) diluted in 0.5% BSA in PBS for 30 minutes at 4°C. Cells were analyzed in a LSR Fortessa cytometer using the FlowJo Software.
In vivo experiments
NOD/SCID (NOD.CB17-Prkdcscid/J) mice (Jackson Laboratory, Bar Harbor, Maine) were provided by the animal facility at the State University of Campinas (CEMIB, UNICAMP, Brazil). The NSGS (NOD.Cg-PrkdcscidIl2rgtm1WjlTg(CMV-IL3,CSF2,KITLG)1Eav/MloySzJ) mice (Jackson Laboratory) were provided by the Boldrini’s animal facility. Ten million IGFBP7 knockdown or Scramble ALL cell lines were injected via the tail vein into nonirradiated NOD/SCID mice. Blood from the retro-orbital plexus was collected weekly to monitor by flow cytometry the percentage of leukemia cells (cells positive for hCD45-FITC, clone HI30; Becton Dickinson) in total CD45+ mononuclear cells: sum of hCD45+ and mCD45+ (mCD45-PE, clone 30F-11; Becton Dickinson). After 4 or 6 weeks, mice were killed, and blood, spleen, liver, and bone marrow were collected to evaluate the percentage leukemia cells. For survival analyses, animals were killed in the moribund state, and Kaplan-Meier curves were compared using the log-rank test. The therapeutic efficacy of C311 anti-IGFBP7 vs polyclonal Balb/c, given intraperitoneally, at 25 µg 3 times per week for 4 weeks was tested against a T-cell ALL (T-ALL, patient T979) xenograft in nonirradiated NSGS mice. Treatment initiated 24 hours after transplantation. In another experiment, using a BCP-ALL (patient B1421) and nonirradiated NOD/SCID, treatment awaited overt leukemia (≥0.5% hCD45 cells in the peripheral blood of half of the animals), and consisted of 20 µg of the C311 anti-IGFBP7 or an irrelevant anti-PSA (Rheabiotech) antibody, intraperitoneally, 3 times per week for 4 weeks. The percentage of ALL cells in peripheral blood mononuclear cells was monitored weekly. The study was approved by the Animal Experimentation Ethics Committees of the State University of Campinas (CEUA/UNICAMP, protocol 1133/2008) and Centro Infantil Boldrini (CEUA/BOLDRINI, protocol 0006/2020).
Results
IGFBP7 knockdown or neutralization decrease the proliferation, survival, and migration of ALL cell lines
To determine whether IGFBP7 has an autocrine role in ALL, different BCP- and T-ALL cell lines were stably transduced with lentiviral particles carrying shRNA constructs directed against IGFBP7 (sh.959 and sh.812) or a noncoding random sequence (sh.Scramble). Downregulation of secreted IGFBP7 levels was confirmed by ELISA and Western blot (supplemental Figure 1A-B). IGFBP7 knockdown markedly reduced the growth rate of sh.IGFBP7 cells in comparison with sh.Scramble-transduced controls (Figure 1A; supplemental Figure 1C), confirming our previous findings on the mitogenic effect of IGFBP7 in BCP-ALL cell lines.9 Accordingly, BrdU incorporation assays revealed a decreased rate of nucleotide incorporation during S-phase on IGFBP7 downregulation, accompanied by rising of the sub-G1/G0 population of apoptotic cells (Figure 1B; supplemental Figure 2A). The deleterious effect of IGFBP7 knockdown on cell viability was confirmed by culturing sh.959 or control cells with suboptimal (3%) serum concentration (Figure 1C; supplemental Figure 2B-C).
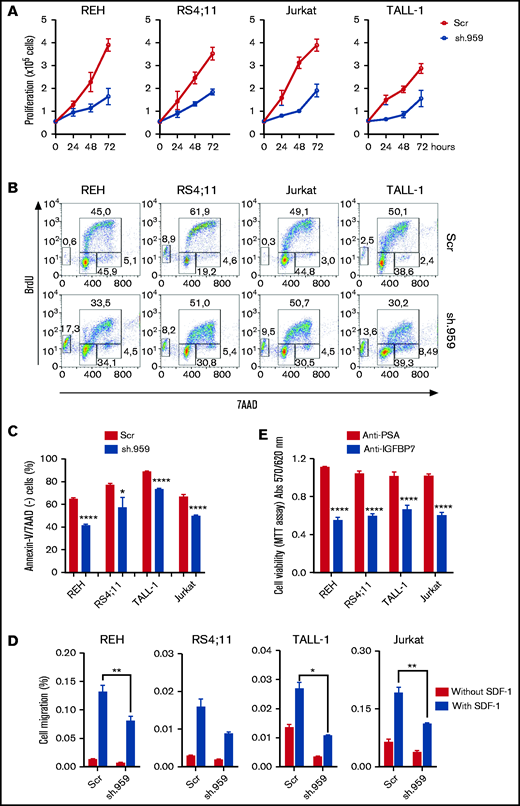
IGFBP7 knockdown or neutralization decreases ALL cell line proliferation, survival, and migration. (A) Proliferation of REH, RS4;11 (BCP-ALL), TALL-1, and Jurkat (T-ALL) cell lines stably transduced with lentivirus expressing shRNA against IGFBP7 (sh.959) or scramble control (Scr) analyzed by the trypan blue exclusion technique. Results from 3 independent experiments run in triplicate wells, using RPMI-3% FBS. (B) Cell cycle analysis of cells after 4-hour incubation with BrdU, using RPMI-10% FBS. The percentage of cells in G0/G1, S-phase, G2/M, and in apoptosis (sub-G0/G1) was evaluated by flow cytometry. Results representative of 3 independent experiments run in triplicate wells (see supplemental Figure 2A). (C) Cells were cultured in RPMI-3% FBS for 24 hours, and the percentage of apoptotic cells was evaluated by flow cytometry after Annexin-V-FITC/7AAD staining. Bars represent means ± standard error (SE) of 4 independent experiments run in duplicate wells. See also supplemental Figure 2C. (D) Migration of ALL cells in the transwell system toward SDF-1 (100 ng/mL) or PBS 1×, using RPMI-10% FBS. Cells in the lower chamber were counted by flow cytometry after 4-hour migration. Bars represent means ± SE for triplicate wells. (E) Viability of ALL cells measured by the MTT assay after 48-hour culture in RPMI-10% FBS supplemented with an anti-IGFBP7 (clone C311, 20 µg/mL) or anti-PSA control (20 µg/mL) monoclonal antibody. Bars represent means ± SE for 3 independent experiments run in triplicate wells. Statistical analyses were done by 1- or 2-way analysis of variance (ANOVA) and Bonferroni posttests (*P ≤ .05, **P .01, ***P .001, and ****P .0001).
IGFBP7 knockdown or neutralization decreases ALL cell line proliferation, survival, and migration. (A) Proliferation of REH, RS4;11 (BCP-ALL), TALL-1, and Jurkat (T-ALL) cell lines stably transduced with lentivirus expressing shRNA against IGFBP7 (sh.959) or scramble control (Scr) analyzed by the trypan blue exclusion technique. Results from 3 independent experiments run in triplicate wells, using RPMI-3% FBS. (B) Cell cycle analysis of cells after 4-hour incubation with BrdU, using RPMI-10% FBS. The percentage of cells in G0/G1, S-phase, G2/M, and in apoptosis (sub-G0/G1) was evaluated by flow cytometry. Results representative of 3 independent experiments run in triplicate wells (see supplemental Figure 2A). (C) Cells were cultured in RPMI-3% FBS for 24 hours, and the percentage of apoptotic cells was evaluated by flow cytometry after Annexin-V-FITC/7AAD staining. Bars represent means ± standard error (SE) of 4 independent experiments run in duplicate wells. See also supplemental Figure 2C. (D) Migration of ALL cells in the transwell system toward SDF-1 (100 ng/mL) or PBS 1×, using RPMI-10% FBS. Cells in the lower chamber were counted by flow cytometry after 4-hour migration. Bars represent means ± SE for triplicate wells. (E) Viability of ALL cells measured by the MTT assay after 48-hour culture in RPMI-10% FBS supplemented with an anti-IGFBP7 (clone C311, 20 µg/mL) or anti-PSA control (20 µg/mL) monoclonal antibody. Bars represent means ± SE for 3 independent experiments run in triplicate wells. Statistical analyses were done by 1- or 2-way analysis of variance (ANOVA) and Bonferroni posttests (*P ≤ .05, **P .01, ***P .001, and ****P .0001).
Chemokines and cell migration play a key role in establishing the leukemia bone marrow niche and in organs infiltration on disease progression. IGFBP7 knockdown significantly reduced the migration of ALL cells in the transwell system toward SDF-1 (Figure 1D). Being an extracellular protein, IGFBP7 is an attractive target for therapeutic intervention using monoclonal antibodies. We produced an anti-IGFBP7 antibody (clone C311) and tested its effects on ALL cell viability. Both our own antibody (Figure 1E) and a commercial one (supplemental Figure 2D) significantly reduced ALL cell viability when added to the culture medium, whereas no effect was seen with an anti-PSA control.
IGFBP7 potentiates the insulin prosurvival effect on primary ALL cells
After confirming by orthogonal approaches that IGFBP7 exerts an autocrine mitogenic and prosurvival effect in ALL, we investigated whether this was dependent on the presence of its ligand insulin. As shown in Figure 2A-B, IGFBP7 mitogenic action was clearly dependent on the combined addition of insulin. Although IGFBP7 or insulin alone promoted cell proliferation in half of the samples tested, their effect was clearly higher and more frequent when used in combination (supplemental Figure 3).

The prosurvival effect of IGFBP7 is insulin (IGF) dependent. (A) ALL cell lines were cultured in RPMI-10% FBS supplemented with insulin (500 ng/mL) and/or IGFBP7 (100 ng/mL) for 48 hours. Cell viability was quantified by the MTT assay. Bars represent means ± SE for 2 independent experiments run in triplicate wells. (B) Proliferation of RS4;11 and Jurkat cell lines expressing shRNAs against IGFBP7 (sh.959) or scramble control (Scr) analyzed by the trypan blue exclusion technique after insulin (500 ng/mL) and/or IGFBP7 (100 ng/mL) treatment. Results from a single experiment run in triplicate wells, using RPMI-3% FBS. See also supplemental Figure 3. (C) Primary BCP-ALL cells after 48 hours or (D) primary T-ALL cells after 24 hours cultured in AIM-V serum-free medium supplemented with insulin (500 ng/mL) and/or IGFBP7 (100 ng/mL). Cell viability was measured by Annexin-V/7AAD staining and flow cytometry. Bars represent means ± SE of Annexin-V/7AAD negative fraction of 2 independent experiments run in duplicate wells. See also supplemental Figure 4A. (E) Primary BCP or T-ALL cells were cultured in AIM-V serum-free medium (Control) supplemented with an anti-PSA control (20 µg/mL) or anti-IGFBP7 antibody (clone C311, 20 µg/mL) for 48 or 24 hours, respectively. Apoptosis was measured by Annexin-V/7AAD staining and flow cytometry. See also supplemental Figure 4B. High IGFBP7 concentrations (20 µg/mL) are detrimental to primary BCP- and T-ALL (F) cells after 24 hours of treatment in AIM-V serum-free medium supplemented with insulin (500 ng/mL). See also supplemental Figure 4C. Statistical analysis was done by 1- or 2-way ANOVA and Bonferroni posttests (*P .05, **P .01, ***P .001, and ****P .0001).
The prosurvival effect of IGFBP7 is insulin (IGF) dependent. (A) ALL cell lines were cultured in RPMI-10% FBS supplemented with insulin (500 ng/mL) and/or IGFBP7 (100 ng/mL) for 48 hours. Cell viability was quantified by the MTT assay. Bars represent means ± SE for 2 independent experiments run in triplicate wells. (B) Proliferation of RS4;11 and Jurkat cell lines expressing shRNAs against IGFBP7 (sh.959) or scramble control (Scr) analyzed by the trypan blue exclusion technique after insulin (500 ng/mL) and/or IGFBP7 (100 ng/mL) treatment. Results from a single experiment run in triplicate wells, using RPMI-3% FBS. See also supplemental Figure 3. (C) Primary BCP-ALL cells after 48 hours or (D) primary T-ALL cells after 24 hours cultured in AIM-V serum-free medium supplemented with insulin (500 ng/mL) and/or IGFBP7 (100 ng/mL). Cell viability was measured by Annexin-V/7AAD staining and flow cytometry. Bars represent means ± SE of Annexin-V/7AAD negative fraction of 2 independent experiments run in duplicate wells. See also supplemental Figure 4A. (E) Primary BCP or T-ALL cells were cultured in AIM-V serum-free medium (Control) supplemented with an anti-PSA control (20 µg/mL) or anti-IGFBP7 antibody (clone C311, 20 µg/mL) for 48 or 24 hours, respectively. Apoptosis was measured by Annexin-V/7AAD staining and flow cytometry. See also supplemental Figure 4B. High IGFBP7 concentrations (20 µg/mL) are detrimental to primary BCP- and T-ALL (F) cells after 24 hours of treatment in AIM-V serum-free medium supplemented with insulin (500 ng/mL). See also supplemental Figure 4C. Statistical analysis was done by 1- or 2-way ANOVA and Bonferroni posttests (*P .05, **P .01, ***P .001, and ****P .0001).
Because cell lines are not good surrogates for primary ALL cells, some experiments were repeated using short-term culture of primary BCP- and T-ALL cells. As shown in Figure 2C-D (supplemental Figure 4A), the combined addition of IGFBP7 and insulin protected primary ALL cells from apoptosis, as visualized by the increased number of Annexin-V/7AAD–negative cells. Likewise, addition of anti-IGFBP7 antibody (clone C311) resulted in a drastic reduction of cell viability (Figure 2E; supplemental Figure 4B). No clear association could be found between the survival of ALL cells and the corresponding mRNA expression levels of INSR, IGF1R, or IGFBP7 (supplemental Figure 5A-B). Of note, these experiments were performed using physiologic levels of IGFBP7, which in the diagnostic bone marrow plasma from children with ALL is ∼50 ng/mL.9 When nonphysiologic, much higher concentrations were used (20 μg/mL) to reproduce some of our fellows’ contradictory results11,12 IGFBP7 caused an inhibitory effect on primary ALL cell viability (Figure 2F; supplemental Figure 4C).
IGFBP7 prolongs IGF1R but not INSR activation in primary ALL cells
Insulin/IGFs act by binding to homo- or heterodimeric INSR and IGF1R cell surface tyrosine kinase receptors that recruit and phosphorylate insulin receptor substrate proteins (IRS1-4), thus initiating the downstream activation of the phosphatidylinositol 3-kinase/Akt/mTOR and Ras/Raf/MAPK signaling pathways.2 Here we found that IGFBP7 significantly enhanced IRS-1 (pan-tyrosine) phosphorylation on insulin stimulation (Figure 3A). Interestingly, treatment of ALL cell lines with IGFBP7 plus insulin resulted in increased IRS-1 phosphorylation for at least 4 hours but not when these factors were added separately. Likewise, IGFBP7 plus insulin prolonged Akt (S473) and Erk1/2 phosphorylation in IGFBP7 knockdown cell lines (Figure 3B). Sustained Akt (S473) phosphorylation by the combined stimulation of cells with IGFBP7 plus insulin was confirmed in 18 primary ALL samples analyzed (Figure 4A; supplemental Figure 6; supplemental Table 1). When the status of IGF1Rβ (Tyr1131) and INSRβ (Tyr1150/1151) was addressed, both showed similar levels of activation at the 15-minute time point and both for the insulin and IGFBP7 plus insulin treatments. At the 4-hour time point, however, only the IGF1Rβ (Tyr1131) remained phosphorylated and exclusively when primary ALL cells were treated with the IGFBP7 plus insulin combination. Insulin treatment alone was not sufficient to keep the receptor active (Figure 4B-C; supplemental Figures 7 and 8). Similar results were obtained with the use of IGF1; however, doses 5 times lower than insulin were required, because 5 ng/mL IGF1 resulted in IGF1Rβ (Tyr1131) phosphorylation at levels equal to those obtained with 25 ng/mL of insulin (Figure 4D). Two conclusions could be drawn from these findings: first, IGFBP7 did not simply increase the half-life of insulin,2 otherwise both IGF1R and INSR should have been activated at the 4-hour time point; and second, the IGF1R seemed to be the candidate molecule in mediating the IGFBP7 potentiation of insulin/IGF1 stimulus. To picture the importance of IGF1R in ALL, we silenced the IGF1R gene in the REH and Jurkat cell lines using an Isopropyl β-D-1-thiogalactopyranoside (IPTG)-inducible shRNA vector. IGF1R knockdown was strongly detrimental in terms of proliferation and survival of both cell lines (Figure 4E-F; supplemental Figure 9A-B).

IGFBP7 prolongs IRS, AKT, and ERK activation by insulin in ALL cell lines. (A) ELISA results for Phospho-IRS-1 (pan-Tyrosine; #7133, Cell Signaling Technology) on different ALL cell lines that were starved for 4 hours in serum free RPMI-1640 medium and then left untreated (Control) or stimulated for 15 minutes or 4 hours with insulin (500 ng/mL) and/or IGFBP7 (100 ng/mL). Bars represent means ± SE for triplicate wells. (B) Western blot results for Phospho-Akt (Ser473) and Phospho-Erk1/2 on the RS4;11 and TALL-1 ALL cell lines stably expressing a scramble shRNA (Scr) or shRNA against IGFBP7 (sh.959), after 4-hour starvation in serum-free RPIM-1640 and left untreated or stimulated for the indicated time with insulin (500 ng/mL) and/or IGFBP7 (100 ng/mL). The effect of IGFBP7 in prolonging (4 hours) AKT and ERK activation is better visualized in sh.959 cells. Of note, IGFBP7 is not needed for the short-term (15 minutes) stimulation of ALL cells with insulin (supplemental Figure 14). Statistical analysis was done by 2-way ANOVA and Bonferroni posttests (*P .05, **P .01, ***P .001, and ****P .0001).
IGFBP7 prolongs IRS, AKT, and ERK activation by insulin in ALL cell lines. (A) ELISA results for Phospho-IRS-1 (pan-Tyrosine; #7133, Cell Signaling Technology) on different ALL cell lines that were starved for 4 hours in serum free RPMI-1640 medium and then left untreated (Control) or stimulated for 15 minutes or 4 hours with insulin (500 ng/mL) and/or IGFBP7 (100 ng/mL). Bars represent means ± SE for triplicate wells. (B) Western blot results for Phospho-Akt (Ser473) and Phospho-Erk1/2 on the RS4;11 and TALL-1 ALL cell lines stably expressing a scramble shRNA (Scr) or shRNA against IGFBP7 (sh.959), after 4-hour starvation in serum-free RPIM-1640 and left untreated or stimulated for the indicated time with insulin (500 ng/mL) and/or IGFBP7 (100 ng/mL). The effect of IGFBP7 in prolonging (4 hours) AKT and ERK activation is better visualized in sh.959 cells. Of note, IGFBP7 is not needed for the short-term (15 minutes) stimulation of ALL cells with insulin (supplemental Figure 14). Statistical analysis was done by 2-way ANOVA and Bonferroni posttests (*P .05, **P .01, ***P .001, and ****P .0001).
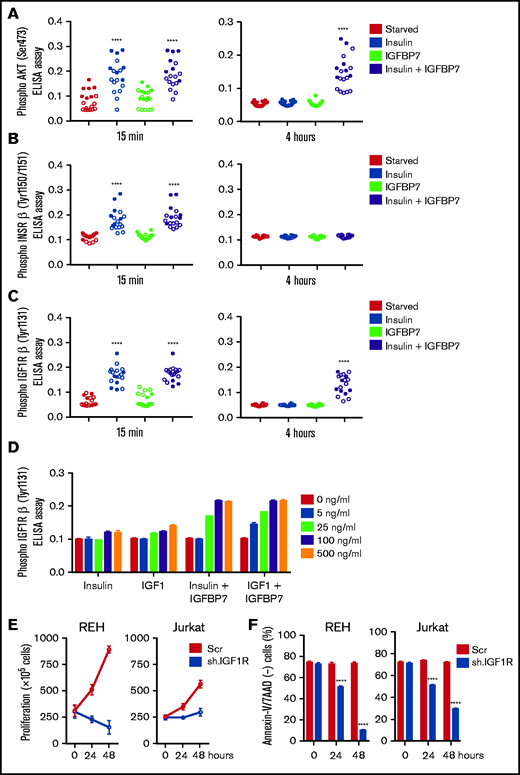
IGFBP7 prolongs IGF1R activation by insulin or IGF1 in primary ALL cells. (A) ELISA results for AKT (Ser473) phosphorylation, (B) INSRβ (Tyr1150/1151) phosphorylation, and (C) IGF1Rβ (Tyr1131) phosphorylation in 11 primary BCP (open circles) and 7 T-ALL (solid circles) cells cultured in AIM-V serum-free medium supplemented with insulin (500 ng/mL), IGFBP7 (100 ng/mL), or a combination of both for 15 minutes and 4 hours. Circles represent means of 2 independent experiments run in duplicate wells. See also supplemental Figures 6, 7, and 8. (D) Dose-response effect for insulin and IGF1, alone or in combination with IGFBP7, on long-term (4 hours) phosphorylation of IGF1Rβ (Tyr1131) as measured by ELISA. Bars represent means ± SE for duplicate wells. (E) Proliferation and (F) survival of REH and Jurkat cell lines stably transduced with IPTG-inducible shRNA vectors against IGF1R or Scramble control (Scr), after 24 and 48 hours of IPTG (1 µg/mL) stimulation, as measured by the trypan blue exclusion technique and Annexin-V/7ADD staining, respectively. Bars represent means ± SE for 2 independent experiments run in triplicate wells. See also supplemental Figure 9A-B. Statistical analysis was done by 1- or 2-way ANOVA and Bonferroni posttests (****P .0001).
IGFBP7 prolongs IGF1R activation by insulin or IGF1 in primary ALL cells. (A) ELISA results for AKT (Ser473) phosphorylation, (B) INSRβ (Tyr1150/1151) phosphorylation, and (C) IGF1Rβ (Tyr1131) phosphorylation in 11 primary BCP (open circles) and 7 T-ALL (solid circles) cells cultured in AIM-V serum-free medium supplemented with insulin (500 ng/mL), IGFBP7 (100 ng/mL), or a combination of both for 15 minutes and 4 hours. Circles represent means of 2 independent experiments run in duplicate wells. See also supplemental Figures 6, 7, and 8. (D) Dose-response effect for insulin and IGF1, alone or in combination with IGFBP7, on long-term (4 hours) phosphorylation of IGF1Rβ (Tyr1131) as measured by ELISA. Bars represent means ± SE for duplicate wells. (E) Proliferation and (F) survival of REH and Jurkat cell lines stably transduced with IPTG-inducible shRNA vectors against IGF1R or Scramble control (Scr), after 24 and 48 hours of IPTG (1 µg/mL) stimulation, as measured by the trypan blue exclusion technique and Annexin-V/7ADD staining, respectively. Bars represent means ± SE for 2 independent experiments run in triplicate wells. See also supplemental Figure 9A-B. Statistical analysis was done by 1- or 2-way ANOVA and Bonferroni posttests (****P .0001).
A previous work has shown that the N-terminal 97-amino-acid portion of IGFBP7 binds to the extracellular portion of IGF1R and suppresses its internalization in response to IGF1.11 Here we confirmed these findings both in ALL cell lines and primary ALL cells (Figure 5A-B; supplemental Figure 10). As expected, insulin treatment of serum-starved ALL cells resulted in significant internalization of both the INSR and IGF1R receptors. When insulin was added in conjunction with IGFBP7, however, only the INSR was internalized, whereas IGF1R remained at the cell surface for as long as the 4 hours tested. As expected, preincubation of ALL cells with the anti-IGFBP7 antibody (clone C311) abolished IGFBP7-induced IGF1R retention at the cell surface. Treating of ALL cells with primaquine or cycloheximide did not interfere with IGFBP7-mediated cell surface retention of IGF1R (Figure 6A-B), suggesting that receptor recycling or synthesis did not contribute to the surface maintenance of IGF1R. Interestingly, treating of IGFBP7-silenced cell lines with the endocytosis inhibitor dansylcadaverine partially restored their proliferative response to insulin (Figure 6C-D). These results are consistent with the notion that IGFBP7 exerts its effects through binding and stabilization of the IGF1R receptor at the surface of ALL cells, thus prolonging its response to insulin.
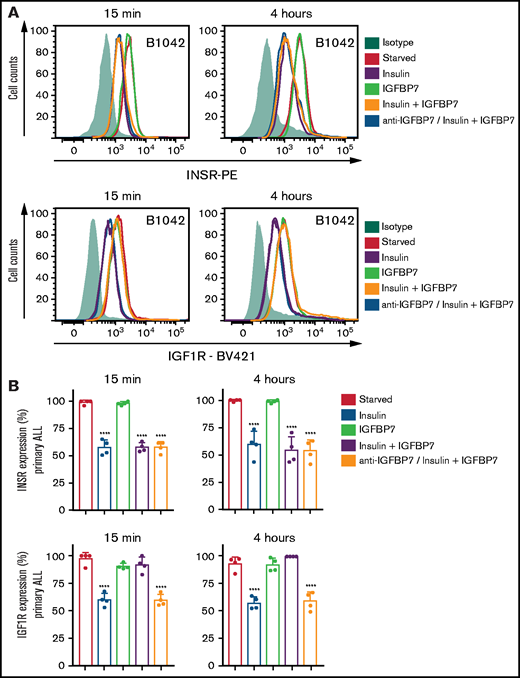
IGFBP7 inhibits IGF1R internalization on insulin stimulation. (A) Cell surface expression of the INSR and IGF1R in a representative case of serum-starved primary BCP-ALL (B1042) cells after 15 minutes or 4 hours of treatment with insulin (500 ng/mL), IGFBP7 (100 ng/mL), anti-IGFBP7 (clone C311, 20 µg/mL), or associations, as measured by flow cytometry analysis. The anti-IGFBP7 antibody (clone C311) was added 30 minutes before insulin/IGFBP7 addition. (B) Normalized results from 4 different primary BCP-ALL (B1042 and B1421) and T-ALL (T1238 and T1260) cells, according to the times and treatments described above. A histogram overlay was used to calculate the percentage of labeled cells with respect to cells left untreated (Starved). See also supplemental Figure 10. Statistical analysis was done by 1-way ANOVA and Bonferroni posttests (****P .0001).
IGFBP7 inhibits IGF1R internalization on insulin stimulation. (A) Cell surface expression of the INSR and IGF1R in a representative case of serum-starved primary BCP-ALL (B1042) cells after 15 minutes or 4 hours of treatment with insulin (500 ng/mL), IGFBP7 (100 ng/mL), anti-IGFBP7 (clone C311, 20 µg/mL), or associations, as measured by flow cytometry analysis. The anti-IGFBP7 antibody (clone C311) was added 30 minutes before insulin/IGFBP7 addition. (B) Normalized results from 4 different primary BCP-ALL (B1042 and B1421) and T-ALL (T1238 and T1260) cells, according to the times and treatments described above. A histogram overlay was used to calculate the percentage of labeled cells with respect to cells left untreated (Starved). See also supplemental Figure 10. Statistical analysis was done by 1-way ANOVA and Bonferroni posttests (****P .0001).

Receptor recycling or synthesis do not contribute to IGF1R retention at the cell surface on IGFBP7 treatment. RS4;11 or Jurkat cells were starved for 4 hours in serum-free RPMI medium and then left untreated or stimulated for 4 hours with insulin (500 ng/mL), IGFBP7 (100 ng/mL), and their combination. Receptor recycling, synthesis, and endocytosis were inhibited by concomitant addition of (A) primaquine (100 µM), (B) cycloheximide (20 µM), or (C) dansylcadaverine (DC; 10 µg/mL), respectively. The cell surface expression of IGF1R was measured by flow cytometry. (D) Proliferation of RS4;11 and Jurkat cells expressing shRNAs against IGFBP7 (sh.959) or scramble control (Scr) analyzed by the trypan blue exclusion technique after treatment with insulin (500 ng/mL), dansylcadaverine (DC; 10 µg/mL), or their combination. Results from a single experiment run in triplicate wells, using RPMI-3% FBS.
Receptor recycling or synthesis do not contribute to IGF1R retention at the cell surface on IGFBP7 treatment. RS4;11 or Jurkat cells were starved for 4 hours in serum-free RPMI medium and then left untreated or stimulated for 4 hours with insulin (500 ng/mL), IGFBP7 (100 ng/mL), and their combination. Receptor recycling, synthesis, and endocytosis were inhibited by concomitant addition of (A) primaquine (100 µM), (B) cycloheximide (20 µM), or (C) dansylcadaverine (DC; 10 µg/mL), respectively. The cell surface expression of IGF1R was measured by flow cytometry. (D) Proliferation of RS4;11 and Jurkat cells expressing shRNAs against IGFBP7 (sh.959) or scramble control (Scr) analyzed by the trypan blue exclusion technique after treatment with insulin (500 ng/mL), dansylcadaverine (DC; 10 µg/mL), or their combination. Results from a single experiment run in triplicate wells, using RPMI-3% FBS.
IGFBP7 knockdown or neutralization decreases the progression of ALL in vivo
Considering the complexity of the insulin/IGF system, in vitro experiments are just a poor approximation for the real situation. For instance, several proteases are known to cleave IGFBPs/IGFBP-rPs, liberating insulin/IGF for INSR/IGF1R binding. In addition, interaction of IGFBPs/IGFBP-rPs with extracellular matrix components can modulate their affinity for insulin/IGFs.13 To address the leukemogenic potential of autocrine IGFBP7 secreted by ALL cells, under physiologic paracrine/endocrine IGFBP7 levels and under the interaction/competition with other cells and molecules, we transplanted the sh.959 cell lines into immunocompromised NOD/SCID mice. Silencing of IGFBP7 resulted in significant attenuation of leukemia progression, as evaluated by the percentage of leukemia cells (human CD45+) in the peripheral blood, bone marrow, liver, and spleen, in relation to mice transplanted with Scramble cells (Figure 7A). Moreover, IGFBP7 knockdown resulted in a significant increase in the Kaplan-Meier survival curves of mice (Figure 7B). Similar results were obtained in mice transplanted with patient-derived xenografts (PDX-ALL; Figure 7C-D) or the RS4;11 cell line (Figure 7E). Mice treated with the anti-IGFBP7 antibody (clone C311) showed decreased leukemic progression and survived longer than animals treated with an isotype or polyclonal antibody controls. Of note, the mouse ortholog of IGFBP7 is functional on human IGF1R (supplemental Figure 11), and our anti-IGFBP7 antibody binds both human and mouse IGFBP7 (supplemental Figure 2E).
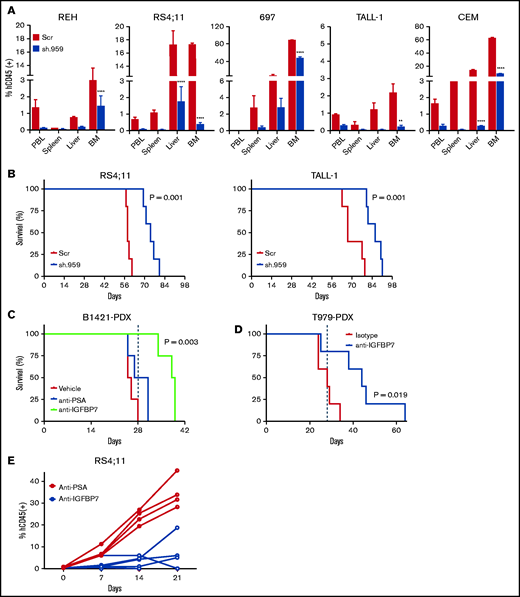
IGFBP7 knockdown or neutralization decreases the progression of ALL in vivo. (A) NOD/SCID mice were transplanted with 10 million sh.959 or Scramble (Scr) ALL cell lines. After 4 (REH, RS4;11, and 697) to 6 (TALL-1 and CCRF-CEM) weeks, animals were killed and evaluated for the percentage of leukemia (hCD45+) cells in the peripheral blood (PBL), spleen, liver, and bone marrow (BM) by flow cytometry. Bars represent means ± SE for 3 animals. Statistical analysis was done by 2-way ANOVA and Bonferroni posttests (**P ≤ .01 and ****P ≤ .0001). (B) Kaplan-Meier survival curves of NOD/SCID mice (5 animals per group) transplanted with sh.959 or Scramble (Scr) ALL cells. (C) Survival curves of NOD/SCID mice transplanted with a patient-derived BCP-ALL xenograft (B1421-PDX) and treated with the anti-IGFBP7 (clone C311) or anti-PSA antibody, as above, for 4 weeks. Treatment of mice (4 animals per group) started when half of the animals had ≥0.5% of leukemia cells
IGFBP7 knockdown or neutralization decreases the progression of ALL in vivo. (A) NOD/SCID mice were transplanted with 10 million sh.959 or Scramble (Scr) ALL cell lines. After 4 (REH, RS4;11, and 697) to 6 (TALL-1 and CCRF-CEM) weeks, animals were killed and evaluated for the percentage of leukemia (hCD45+) cells in the peripheral blood (PBL), spleen, liver, and bone marrow (BM) by flow cytometry. Bars represent means ± SE for 3 animals. Statistical analysis was done by 2-way ANOVA and Bonferroni posttests (**P ≤ .01 and ****P ≤ .0001). (B) Kaplan-Meier survival curves of NOD/SCID mice (5 animals per group) transplanted with sh.959 or Scramble (Scr) ALL cells. (C) Survival curves of NOD/SCID mice transplanted with a patient-derived BCP-ALL xenograft (B1421-PDX) and treated with the anti-IGFBP7 (clone C311) or anti-PSA antibody, as above, for 4 weeks. Treatment of mice (4 animals per group) started when half of the animals had ≥0.5% of leukemia cells
Discussion
Although insulin and IGF1 have been known to enhance ALL survival/proliferation in vitro for decades,14-16 their role in ALL has not been fully explored, except in T-ALL where IGF1R expression was shown to be under Notch1 control and to play a fundamental role in T-ALL progression and transplantability in mice.17,18 Interestingly, tumor-associated dendritic cells support T-ALL growth via IGF1R activation.19 Our gene expression data indicate that primary BCP-ALL and T-ALL express both the INSR and IGF1R, although INSR seemed lower in T-ALL (supplemental Figure 12B). This was confirmed by us (Figure 5A; supplemental Figure 10) and others5,18 by flow cytometry analyses. Thus, ALL cells seem ready to respond to insulin/IGFs. However, as we showed here, addition of insulin/IGFs alone has a rather small effect on ALL survival (supplemental Figure 13). Association of IGFBP7 with the insulin/IGF stimulus seemed mandatory to have any significant effect on primary ALL survival, and this may be the reason why the role of insulin/IGF1 in ALL biology could have been underappreciated in previous studies.
High IGFBP7 expression has been associated with treatment resistance20 and/or worse prognosis in ALL.9,21,22 Here, we demonstrate that IGFBP7 exerts an autocrine prosurvival and mitogenic effect on ALL that is dependent on insulin/IGFs. Results were confirmed in vivo. By using IGFBP7 knockdown ALL cell lines or antibody-mediated neutralization of IGFBP7 in animals transplanted with primary ALL, we demonstrated the relevance of autocrine or extracellularly available IGFBP7 levels, respectively, in ALL fitness, at physiologic concentrations of insulin, IGFs, INSR, IGF1R, IGFBPs, extracellular matrix components, and all other factors that modulate insulin/IGF action.4
On activation and autophosphorylation, receptor tyrosine kinases undergo rapid internalization, mainly by clathrin-mediated endocytosis.23 The INSR has a C-terminal motif for MAD2 binding, which in turn recruits BUBR1 and the clathrin adaptor protein complex AP2, which facilitates clathrin coating and endocytosis on receptor activation.24 Conversely, IGF1R has no such MAD2 binding motif and exhibits prolonged perdurance at the cell surface, apparently mediated by IRS-1 binding to and blocking of AP2.25 In keeping with previous findings in breast cancer,11 we found that IGFBP7 inhibited IGF1R internalization despite insulin stimulation. The cell surface retention of IGF1R in ALL was found to prolong the insulin-induced phosphorylation of IGF1Rβ, IRS-1, ERK, and AKT. In contrast, in breast cancer MCF10A and MCF10CA1a cell lines, the cell surface–retained IGF1R was insensitive to IGF1 or insulin. Hypothetically, IGFBP7 binding to unoccupied IGF1R sterically restricts or allosterically prevents subsequent binding of IGF1/insulin.11 However, this inhibitory effect required preincubation of breast cancer cells for 2 hours with IGFBP7 before addition of growth factors. Simultaneous addition of IGFBP7 with insulin/IGF1 had no effect.11 Here, IGFBP7 and insulin or IGF1 was added simultaneously. Although differences in the INSR/IGF1R signaling and/or internalization complexes between ALL and breast cancer cells should not be undervalued, we suspect that the amount of IGFBP7 added to cells likely contributed to the discrepancy. Evdokimova et al11 used IGFBP7 at 20 μg/mL, whereas we used 100 ng/mL. At 20 μg/mL, we also found an inhibitory effect of IGFBP7 on ALL cells. It is plausible that at this high concentration, IGFBP7 may indeed restrict or prevent IGF1/insulin binding to the IGF1R, but 20 μg/mL is far above the physiologic levels of IGFBP7 in adult serum (21-35 ng/mL)26 or childhood ALL bone marrow plasma (49 ng/mL).9 Unfortunately, the use of supraphysiologic amounts of IGFBP7 has been the rule rather than the exception in the literature. As shown here, physiologic levels of IGFBP7 promoted the perdurance of IGF1R at the surface of ALL cells, prolonging insulin/IGFs stimulation. Accordingly, a recent study in mouse hepatocytes showed that IGFBP7 (20 ng/mL) binds to the INSR, potentiating its activation by insulin.27 Likewise, ectopic expression of IGFBP7 in the breast cancer MDA-MB468 cell line enhanced IGF1Rβ/INSRβ phosphorylation by insulin,11 an effect probably resulting from IGF1Rβ activation because this cell line expresses very low levels of INSR.28
ALL cells express both the INSR (INSR-A isoform) and the IGF1R (supplemental Figure 12A-B). INSR and IGF1R form hybrid receptors consisting of one molecule of INSR and one molecule of IGF1R. Homotypic INSR and IGF1R have the strongest binding affinity for insulin and IGF1, respectively, whereas INSR/IGF1R hybrid receptors display high affinity binding for both insulin and IGF1.29 At the supraphysiologic levels (500 ng/mL or 100 nM) used in this study, insulin was expected to bind INSR, IGF1R, and INSR/IGF1R receptors. However, only IGF1R was retained at the cell surface and showed prolonged phosphorylation on IGFBP7/insulin treatment of ALL cells. Previous coimmunoprecipitation assays demonstrated that IGFBP7 binds the extracellular part of IGF1R but not INSR.11 Thus, we deduce that IGFBP7 exerted its effect by binding to homotypic IGF1R. Otherwise, we would expect the INSR chain, at least from heterotypic INSR/IGF1R receptors, to be phosphorylated as well.
Insulin is at least 200-fold less specific for the IGF1R than is IGF1.29 Interestingly, monoclonal antibodies directed against the extracellular part of IGF1R are able to dramatically increase insulin binding to homotypic IGF1R to affinity levels approaching IGF1 binding. Apparently, these antibodies could promote insulin binding by inducing a conformational change in the IGF1R, which results in loss of inhibitory constraints.30 We speculate whether something similar was produced by IGFBP7, because 25 ng/mL insulin was able to activate the IGF1R to levels equivalent to those obtained with 5 ng/mL IGF1 (ie, a fivefold difference only).
Different drugs and antibodies targeting IGF1R have been developed, but most did not advance to clinical trials.31 The similarity between IGF1R and INSR receptors and their heterodimerization has been a challenge to the design of specific therapeutic drugs targeting IGF1R.3 The SCH 717454 monoclonal antibody against IGF1R has shown promising in vivo results against 2 of 8 ALL samples tested.31 Here we present IGFBP7 as a new target candidate for therapeutic antibodies directed to modulate the insulin/IGF system. We showed that IGFBP7 neutralization using a monoclonal antibody was safe (in mice) and resulted in significant increments on the survival of mice transplanted with patient-derived xenografts. Although dose-response experiments were not performed, the anti-IGFBP7 antibody was used at 1 mg/kg, every 3 days, which was compatible to that of a recent preclinical study with an Fc-engineered CD19 antibody.32
In conclusion, we confirmed the notion that insulin/IGFs are important mitogenic and prosurvival factors in ALL and revealed IGFBP7 as a relevant player in this context and as a valid target for therapeutic intervention in the treatment of leukemia and possibly other cancers that involve the IGF system.
Acknowledgments
The authors thank Maria Carolina Spago and Marcia Cristina Fornazim, from the State University of Campinas, for excellent technical help with cell culture experiments. L.L.A., A.B.A.L., L.W.C., J.R.C., and P.P.Z. received a scholarship from Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP). J.A.Y. received a productivity fellowship from the Brazilian National Counsel of Technological and Scientific Development (CNPq, process 305896/2013-0 and 301596/2017-4). This work was supported by grants to J.A.Y. from CNPq (471003/2013-1) and FAPESP (12/12802-1 and 14/20015-5).
Authorship
Contribution: L.L.A., A.B.A.L., and J.A.Y. conceived and designed the study; L.L.A. and A.B.A.L. performed all experiments; L.W.C., J.B.C.C., and S.R.B. performed cellular signal transduction analyses by Western blot; J.R.C. performed fluorescence-activated cell sorting analysis; P.P.Z. performed antibody production and PDX animal experiment; A.E.N. contributed analytical tools; S.R.B. contributed patient’s samples and the corresponding clinical data; and L.L.A., A.B.A.L., and J.A.Y. performed statistical analysis and wrote the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: José Andrés Yunes, 1270 Dr. Gabriel Porto St, Campinas, SP 13083-210, Brazil; e-mail: andres@boldrini.org.br.

