

Key Points
JAK1 R629_S632delinsSA is a novel somatic mutation leading to IL-3–independent cell growth, JAK-STAT activation, and clonal eosinophilia.
Functional interrogation of variants of unknown significance detected by NGS panels can uncover oncogenic, druggable targets.
Abstract
Hypereosinophilia (HE) has been defined as persistent eosinophilia >1.5 × 109/L; it is broadly divided into primary HE (clonal or neoplastic; HEN), secondary/reactive HE (HER), or HE of undetermined significance (HEUS) when no cause is identified. The use of myeloid next-generation sequencing (NGS) panels has led to the detection of several mutations in patients previously diagnosed with HEUS, reassigning some patients to the category of HEN, specifically the World Health Organization category of chronic eosinophilic leukemia, not otherwise specified (CEL, NOS). Here, we describe a novel somatic JAK1 pseudokinase domain mutation (R629_S632delinsSA) in a patient with HE that had initially been characterized as a variant of uncertain significance. We performed functional studies that demonstrated that this mutation results in growth factor independence of Ba/F3 cells in vitro and activation of the JAK-STAT pathway. These effects were abrogated by the JAK1/JAK2 inhibitor ruxolitinib. R629_S632delinsSA is the first known somatic mutation in JAK1 linked to a clonal eosinophilic neoplasm, and highlights the importance of the JAK-STAT pathway in eosinophil survival.
Introduction
Hypereosinophilia (HE) is defined as persistent eosinophilia >1.5 ×109/L.1 Chronic eosinophilic leukemia, not otherwise specified (CEL, NOS) is diagnosed in the presence of HE with evidence of a nonspecific clonal cytogenetic or molecular abnormality (eg, exclusion of BCR-ABL1, and fusion tyrosine kinases involving PDGFRA, PDGFRB, FGFR1, or JAK2), and/or an increase in the myeloblast percentage in the marrow or blood <20%.2 With the use of next-generation sequencing (NGS) panels, several otherwise unnoticed pathogenic mutations have been detected in some patients with HE that would lead to the diagnosis of CEL, NOS. Patients with HE in whom mutations are detected by NGS have an inferior prognosis, similar to patients with CEL, NOS.1-4
Activation of the JAK-STAT pathway is critical for eosinophil survival.5 Somatic mutations and structural rearrangements of genes in this pathway have been described in patients with HE. These include JAK2 V617F, JAK2 L583_A586delinsS, STAT3 Y640F, STAT5B N642H, and JAK2 fusion genes.6-9 In contrast, somatic mutations in JAK1 are recurrently seen in T-cell neoplasms and acute lymphoblastic leukemia, but only 1 case has been described in a patient with a myeloproliferative neoplasm (MPN) unclassifiable.10-12 A germline activating JAK1 variant (A634D) has recently been reported in a family with autosomal-dominant hypereosinophilic syndrome.13 Here, we describe a novel somatic JAK1 pseudokinase domain mutation (R629_S632delinsSA), in close proximity to the activating germline variant, in a patient with acquired HE. This mutation results in interleukin-3 (IL-3)–independent growth of Ba/F3 cells in vitro, activation of the JAK-STAT pathway, and sensitivity to ruxolitinib.
Methods
Patient samples
Written informed consent was provided by the patient. The study was approved by the Stanford University Institutional Review Board and was conducted in accordance with the Declaration of Helsinki.
Targeted NGS
NGS was performed on blood using the Stanford Actionable Mutation Panel for Hematopoietic and Lymphoid Malignancies. See supplemental Materials for more information.
Vectors, cloning, cell culture, retrovirus generation, Ba/F3 transformation assays, measurement of drug response by cell proliferation assay, and immunoblotting
JAK1 (NM_002227.2) variant c.1887_1894delinsTG, p.Arg629_Ser632delinsSerAla, and c.1901C>A, p.A634D pDONR vectors were generated in pDONR223-JAK1 vector (Addgene plasmid #23932).14 Standard protocols were used and are detailed in supplemental Materials.
Functional analysis of patient samples
Screening of primary cells against panels of kinase inhibitors was performed as previously described.15 See supplemental Materials for more information
Results and discussion
The patient presented at 17 years of age with a white blood cell (WBC) count of 13.7 × 109/L and a differential count revealing 23% eosinophils (absolute eosinophil count 3.15 × 109/L). She was treated at that time with “an antiparasitic” for Blastocystis hominis in her stools, without a subsequent change in HE. She has since noted intermittent hives with dermographism. She was referred to the Stanford Cancer Institute in 2019, when she was 33 years of age, at which time her physical examination was normal and a complete blood count revealed a WBC of 16.3 × 109/L, hemoglobin of 13.9 g/dL, and platelet count of 256 × 109/L. Manual differential revealed 30% neutrophils, 16% lymphocytes, 3.7% monocytes, 1% basophils, and 29% eosinophils (absolute eosinophil count 4.73 × 109/L). Blood smear revealed an increase in mature eosinophils, with some showing asymmetrical granulation, in addition to hypogranular neutrophils (supplemental Figure 1A). Stool analysis detected B hominis for which she received metronidazole, again without improvement in HE. The infectious and autoimmune work-up was unrevealing (supplemental Materials). The serum tryptase level was elevated at 18.8 ng/mL (normal < 11.5 ng/mL). Testing of the peripheral blood was negative for BCR-ABL1 transcripts by reverse-transcriptase polymerase chain reaction, KIT D816V by allele-specific polymerase chain reaction (sensitivity 0.1%), and a clonal T-cell receptor gene rearrangement by NGS. Marrow evaluation was normocellular (60%) with an increase in mature eosinophils (17%) (supplemental Figure 1B). Immunohistochemical staining with tryptase and CD117 revealed scattered mast cells with no aberrant expression of CD25 or CD2. Cytogenetics revealed a normal female karyotype. Fluorescence in situ hybridization testing did not detect the CHIC2 deletion or rearrangements involving PDGFRB, FGFR1, or JAK2.
NGS of the blood did not reveal pathogenic mutations; however, 3 variants of uncertain significance were identified: an insertion/deletion mutation in the pseudokinase domain of JAK1 (JAK1 R629_S632delinsSA) with a variant allele frequency (VAF) of 23% and 2 substitution mutations in SETD2 (R136T and T2421A) with VAFs of 50% and 47%, respectively. The JAK1 mutation was not detected by NGS in fibroblasts cultured from the patient’s skin, thus confirming it was somatically acquired. In contrast, both SETD2 mutations were detected at a VAF of 50% in the skin cells, confirming their germline origin. We screened our Stanford and Oregon Health & Science University institutional databases for patients who had eosinophilia and a concomitant JAK1 mutation; 1 additional patient was identified (described in supplemental Materials).
We tested the transforming capacity of JAK1 A634D and JAK1 R629_S632delinsSA pseudokinase mutations. Both were capable of inducing transformation of Ba/F3 cells to IL-3–independent growth (starting on day 8 and 3, respectively; Figure 1A). Sanger sequencing did not reveal any additional mutations in JAK1 transgenes from transformed Ba/F3 cells (data not shown). Cells transformed by JAK1 A634D and JAK1 R629_S632delinsSA were sensitive to JAK inhibitors, including ruxolitinib, with 50% inhibitory concentrations (IC50) ∼ 63 nM and 182 nM, respectively (Figure 2A). The JAK1 R629_S632delinsSA-mutant Ba/F3 cells induced high levels of STAT3 and STAT5 phosphorylation, in contrast to parental Ba/F3 cells and Ba/F3 cells harboring wild-type JAK1 (Figure 1B). Similar high levels of STAT3 and STAT5 phosphorylation were seen in JAK1 R629_S632delinsSA–transformed HEK293T cells (Figure 1B). Pretreatment of Ba/F3 cells transformed by each JAK1 mutant with ruxolitinib (250 nM and 500 nM) significantly decreased the phosphorylation of STAT5 (Figure 2B). The patient’s blood and marrow specimens showed partial sensitivity to tofacitinib and JAK inhibitor 1 and were resistant to ruxolitinib (Figure 2C). The variable sensitivity between various JAK inhibitors could be due to the differential inhibition of other JAK kinases (eg, JAK3, TYK2), leading to further inhibition of the JAK/STAT pathway, or an off-target effect. Additionally, the reduced sensitivity of primary blood and marrow cells to ruxolitinib vs transfected Ba/F3 cells could be related to the reduced sensitivity of differentiated leukemic cells to drug-sensitivity assays in vitro.16 In addition, based on mutation VAFs, as well as complete blood count differentials, the neoplastic component of the tested specimens represented only a portion of the total WBCs.
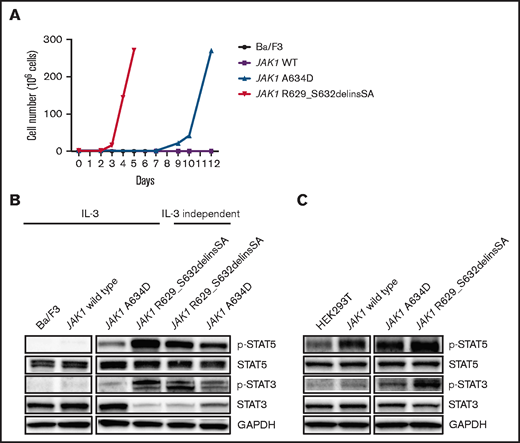
JAK1 pseudokinase mutations demonstrate transforming capability and result in hyperactivation of the JAK/STAT pathway. (A) Growth curves of Ba/F3 cells expressing JAK1 A634D and JAK1 R629_S632delinsSA cultured in the absence of IL-3 (graph is representative of 3 independent experiments). (B) Immunoblot analysis of Ba/F3 parental cells, Ba/F3 cells expressing JAK1 wild-type, JAK1 A634D, and JAK1 R629_S632delinsSA cultured in the presence of IL-3 and IL-3–independent Ba/F3 cells expressing JAK1 A634D, and JAK1 R629_S632delinsSA. (C) Immunoblot analysis of HEK293 cells expressing JAK1 wild type, JAK1 A634D, and JAK1 R629_S632delinsSA. p-, phosphorylated; WT, wild-type.
JAK1 pseudokinase mutations demonstrate transforming capability and result in hyperactivation of the JAK/STAT pathway. (A) Growth curves of Ba/F3 cells expressing JAK1 A634D and JAK1 R629_S632delinsSA cultured in the absence of IL-3 (graph is representative of 3 independent experiments). (B) Immunoblot analysis of Ba/F3 parental cells, Ba/F3 cells expressing JAK1 wild-type, JAK1 A634D, and JAK1 R629_S632delinsSA cultured in the presence of IL-3 and IL-3–independent Ba/F3 cells expressing JAK1 A634D, and JAK1 R629_S632delinsSA. (C) Immunoblot analysis of HEK293 cells expressing JAK1 wild type, JAK1 A634D, and JAK1 R629_S632delinsSA. p-, phosphorylated; WT, wild-type.
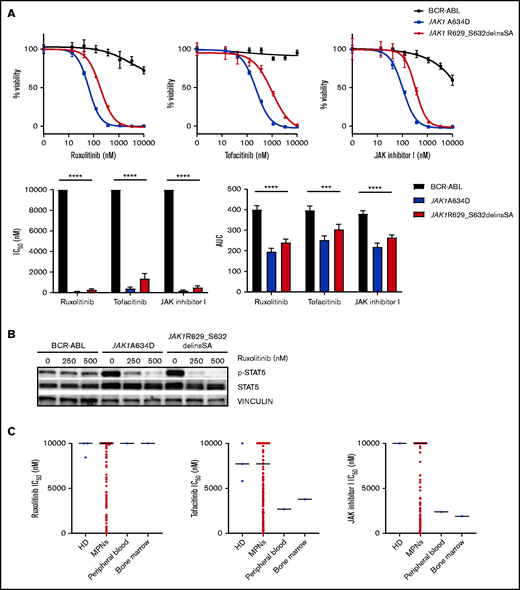
JAK1 pseudokinase mutations demonstrate sensitivity to JAK inhibitors. (A) Representative graphs of the dose-response curves (72-hour sensitivity) for different JAK inhibitors on Ba/F3 cells expressing BCR-ABL, JAK1 A634D, and JAK1 R629_S632delinsSA (upper panels). Graphs showing mean IC50 (lower left panel) and area under the dose-response curve (AUC; lower right panel) for different JAK inhibitors on Ba/F3 cells expressing BCR-ABL, JAK1 A634D, and JAK1 R629_S632delinsSA (3 independent experiments). (B) Immunoblot analysis of IL-3–independent Ba/F3 cells expressing BCR-ABL, JAK1 A634D, and JAK1 R629_S632delinsSA treated with vehicle or 50 nM or 100 nM of ruxolitinib for 4 hours (n = 2 replicates). (C) Sensitivity of peripheral blood and bone marrow specimens from the patient with JAK1 R629_S632delinsSA to JAK inhibitors, represented by IC50, in comparison with control samples obtained from healthy donors (HD) and specimens from a larger cohort of patients with MPNs) (the horizontal lines denote median IC50 of HD and MPN specimens). ****P < .0001, ***P < .001, 1-way analysis of variance. p-, phosphorylated.
JAK1 pseudokinase mutations demonstrate sensitivity to JAK inhibitors. (A) Representative graphs of the dose-response curves (72-hour sensitivity) for different JAK inhibitors on Ba/F3 cells expressing BCR-ABL, JAK1 A634D, and JAK1 R629_S632delinsSA (upper panels). Graphs showing mean IC50 (lower left panel) and area under the dose-response curve (AUC; lower right panel) for different JAK inhibitors on Ba/F3 cells expressing BCR-ABL, JAK1 A634D, and JAK1 R629_S632delinsSA (3 independent experiments). (B) Immunoblot analysis of IL-3–independent Ba/F3 cells expressing BCR-ABL, JAK1 A634D, and JAK1 R629_S632delinsSA treated with vehicle or 50 nM or 100 nM of ruxolitinib for 4 hours (n = 2 replicates). (C) Sensitivity of peripheral blood and bone marrow specimens from the patient with JAK1 R629_S632delinsSA to JAK inhibitors, represented by IC50, in comparison with control samples obtained from healthy donors (HD) and specimens from a larger cohort of patients with MPNs) (the horizontal lines denote median IC50 of HD and MPN specimens). ****P < .0001, ***P < .001, 1-way analysis of variance. p-, phosphorylated.
To our knowledge, this is the first report of a patient with a clonal eosinophilic neoplasm associated with a somatic activating mutation in JAK1. The JAK1 R629_S632delinsSA mutation described herein is in close proximity to a heterozygous germline JAK1 p.A634D mutation described in a family with hypereosinophilic syndrome and somatic acquisition of the same mutation in acute lymphoblastic leukemia.13,17 The germline JAK1 p.A634D mutation activates the JAK-STAT pathway in transfected HEK293 cells, which was dampened by ruxolitinib.13 Furthermore, ruxolitinib treatment of the affected children led to a significant reduction in eosinophilia, as well as resolution of dermatitis and hepatosplenomegaly.13 The variability of the phenotype with different JAK1 activating mutations may relate to the particular JAK1 mutation, the hematopoietic progenitor compartment in which the mutation arises, VAF of the mutation, and/or the presence of modifying comutations and/or epigenetic alternations. The identification in our patient of the somatic JAK1 R629_S632delinsSA mutation, adjacent to the aforementioned germline p.A634D variant, is reminiscent of the discovery of the somatic CSF3R T618I mutation in chronic neutrophilic leukemia,18 which was heralded by a similar germline mutation in a family with hereditary neutrophilia.19
Somatic insertion/deletion mutations (p.L583_A586delinsS) within exon 13 of the pseudokinase domain of JAK2 were described recently in patients with CEL, NOS and a concomitant polycythemia. Transformed Ba/F3 cells were resistant to JAK inhibition in vitro, although ruxolitinib led to a reduction in eosinophil counts in 1 patient.7 JAK1 and JAK2 proteins share a large degree of structural homology (supplemental Figure 1).20-22 This highlights the relevance of the JAK-STAT pathway in the pathogenesis of some eosinophilic neoplasms and the potential role of JAK inhibitors in the treatment of these disorders. Phase 2 clinical trials of the JAK1/JAK2 inhibitor ruxolitinib in primary eosinophilic neoplasms and idiopathic hypereosinophilic syndrome are underway. The patient was not treated with a JAK inhibitor given that she was breastfeeding, lacked clinical symptoms or organ damage, and has maintained a stable eosinophil count for >16 years.
This case illustrates the role of NGS myeloid gene panels in the work-up of patients with idiopathic HE.3 Knowledge of the genes tested in these panels is important because the JAK1 gene is not routinely included in several commercially available NGS panels.23,24 In addition, in many cases evaluated by NGS, variants of uncertain significance annotations simply reflect a lack of data supporting their role in disease pathogenesis. Studies such as the one presented here are needed to better annotate the many uncharacterized somatic variants identified by NGS panels, some of which will be shown to represent oncogenic clinically significant targets that aid in the diagnosis and prognosis of hematologic neoplasms and/or eligibility for targeted therapies. Nevertheless, we cannot rule out that other genetic alterations, not detected by the NGS myeloid gene panel and cytogenetic analysis, might have contributed to the eosinophilic phenotype in this patient.
In summary, we have identified the first somatic mutation in JAK1 associated with clonal eosinophilia. Although such cases are rare, their identification may carry therapeutic importance with the availability of JAK inhibitors.
Acknowledgments
This work was supported in part by the Stanford Cancer Institute (W.S.), National Institutes of Health, National Heart, Lung, and Blood Institute grant 2T32HL120824-060 (W.S.), the Charles and Ann Johnson Foundation (J.G.), the Mark Foundation for Cancer Research (J.W.T.), the Silver Family Foundation (J.W.T.), and National Institutes of Health, National Cancer Institute grants 1R01CA183947, 1U01CA217862, and 1U54CA224019 (J.W.T.).
Authorship
Contribution: W.S., A.D., J.W.T., and J.G. designed the study; A.D., D.S., Q.M., and J.W.T performed experiments; W.S., T.T., F.Y., S.F.-P., R.P., J.Z., and J.G. provided patient data and material; W.S, A.D., T.T., J.T., and J.G. analyzed and interpreted data and wrote the initial draft of the manuscript; and all authors approved the final version of the manuscript.
Conflict-of-interest disclosure: W.S. and J.G. receive funding for the conduct of clinical trials from Incyte. W.S. serves on an advisory board for Incyte. J.G. serves on an advisory board for, and has received honoraria from, Incyte. J.W.T. has received research support from Agios, Aptose, Array, AstraZeneca, Constellation, Genentech, Gilead, Incyte, Janssen, Petra, Seattle Genetics, Syros, Tolero, and Takeda. The remaining authors declare no competing financial interests.
Correspondence: Jason Gotlib, Stanford Cancer Institute, 875 Blake Wilbur Dr, Stanford, CA 94305-6555; e-mail: jason.gotlib@stanford.edu.

